
Control Respiratorio durante el Ejercicio
Karlman Wasserman1, Brian J Whipp1 y Richard Casaburi1
Division of Respiratory Physiology and Medicine, Department of Medicine, Los Angeles County Harbor - UCLA Medical Center, University of California, Los Angeles, Torrance, California.
Artículo publicado en el journal PubliCE, Volumen 0 del año 1994.
Publicado 23 de abril de 2004
INTRODUCCION
Determinar el mecanismo de control de un sistema biológico requiere el conocimiento de las variables que son reguladas. La mayoría de los investigadores están de acuerdo en que la presión parcial del CO2 en la sangre (PaCO2) y el [H+] son normalmente regulados durante el estado de equilibrio (steady-state) de ejercicios de moderada intensidad (77,128,190, 205,226), especialmente en un tranquilo ambiente de laboratorio. Además, a pesar de grandes cambios en la tasa metabólica, los gases sanguíneos arteriales y la homeostasis de pH son mantenidos, a medida que la ventilación cambia adecuadamente para satisfacer los requerimientos del intercambio gaseoso. Si la respuesta ventilatoria fuera inadecuada, la gran tasa de producción de CO2 produciría acidosis respiratoria. Por el contrario, si el aumento ventilatorio fuera excesivo, se produciría alcalosis respiratoria. Ni siquiera ocurre en forma característica el disturbio ácido-base respiratorio. Además, para niveles de ejercicios moderados, la ventilación se combina con el stress metabólico con una precisión remarcable. Por otra parte, tasas intensas de esfuerzo causan una acidosis metabólica como consecuencia del aumento en la producción de ácido láctico. El [H+] producido de esta reacción es un estimulante respiratorio bien reconocido.
Desde que fueron introducidos los sistemas de análisis ventilación-a-ventilación que miden no sólo esta variable sino también el intercambio gaseoso, ha resultado evidente que la respuesta ventilatoria al ejercicio está íntimamente acoplada al intercambio de CO2 (34,53,72,114,226), de manera que aún durante las transiciones de trabajos y pausas de ejercicio, la PaCO2 y el pH permanecen relativamente constantes. Por lo tanto, cualquier hipótesis sobre el control respiratorio durante el ejercicio debe considerar la precisión con la cual son regulados los gases sanguíneos arteriales y el pH. Varias preguntas son, entonces, cruciales: Cuáles son los estímulos específicos?; dónde están los receptores?; cuáles son los mecanismos de mediación? Además, el control se caracteriza por mecanismos de alimentación adelantada que dan paso al intercambio gaseoso requerido para arterializar la sangre venosa, o por un sistema de retroalimentación dependiente de un error captado flujo abajo por un censor influenciado por la composición de la sangre arterial, o por ambos mecanismos?
Este artículo describe el comportamiento del sistema de control respiratorio durante el ejercicio y considera las distintas hipótesis sobre el control ventilatorio. Sin embargo, primero examinaremos algunas de las técnicas de investigación que, durante los últimos 15 años, han permitido tener una visión sobre la hiperpnea en el ejercicio.
NUEVAS TECNICAS PARA ESTUDIAR LA HIPERPNEA EN EL EJERCICIO
El mayor entendimiento sobre el control ventilatorio durante el ejercicio puede ser atribuido principalmente a nuevas técnicas. Por ejemplo, el análisis computado de los datos y el análisis de los sistemas, han expandido, en gran medida, nuestra capacidad para determinar, analizar e interpretar las respuestas al ejercicio.
A) Análisis computado de datos
Las grandes habilidades de las computadoras llevadas a cabo por los programadores, han expandido el límite y mejorado los detalles del estudio de la hiperpnea en el ejercicio. Las computadoras análogas (148,156), e híbridos análogo-digitales (donde los dispositivos análogos realizan el condicionamiento de las señales de inicio), que fueron usados en un principio (9,31,155), fueron reemplazadas por sistemas de computadoras digitales mucho más pequeñas y sofisticadas (19,30,181,203,256).
En un sistema típico, el procesamiento de datos comienza en el momento en el cual los signos eléctricos producidos por los transductores son transformados de análogos a digitales, en una frecuencia donde la onda respiratoria puede ser descripta en una forma adecuada.
Los intervalos de datos de 20 ms son suficientes, aun con una tasa respiratoria de 60 ventilaciones/ min. El muestreo de varios canales de datos requiere de sólo 1 ms, dejando los 19 ms restantes para la computadora, interactuando con el usuario a través de la consola de control y transfiriendo los datos a los dispositivos periféricos. Los resultados de los cálculos realizados por la computadora se pueden guardar en cinta magnética o en discos para su posterior procesamiento. Finalmente, los convertidores digital-análogo pueden presentar gráficamente cualquier serie de cálculos.
Algunos aspectos del análisis computado de las respuestas del ejercicio merecen una mención especial.
CALCULOS VENTILACION-A-VENTILACION. El más importante evento en respiración es el acto de la ventilación. Los cálculos se pueden hacer programando a la computadora para que reconozca el comienzo y el final de las fases de inspiración y espiración de la ventilación.
El flujo ventilatorio, el volumen corriente (VT; a partir de la integración digital del flujo ventilatorio), y la duración de cada ventilación se pueden determinar con gran exactitud. A partir de estas variables, otras también pueden ser calculadas, tal como la ventilación de espiración por minuto (VE), la frecuencia ventilatoria, y la descripción de la forma de la onda del flujo ventilatorio.
CALCULOS DEL INTERCAMBIO GASEOSO. Se han desarrollado analizadores de gases de O2 y CO2 de respuestas muy rápidas. Con estos analizadores es ahora muy práctico medir el intercambio de CO2 y O2 ventilación-a-ventilación [producción de CO2 (VCO2) y consumo de O2 (VO2)] (19,21) por integración digital continua del producto del flujo de aire espirado y la concentración gaseosa fraccional sobre la exhalación. Sin embargo, para que estas determinaciones sean válidas, se necesita tener en cuenta factores tales como el tiempo del tránsito del gas al analizador, y la variación en las presiones del vapor de agua y en la temperatura (19,21). Además, asumiendo una igualdad de intercambio de N2 a nivel alveolar-capilar, uno puede compensar para los cambios ventilación-a-ventilación en las reservas de gases pulmonares, y calcular las tasas reales de intercambio de O2 y CO2 con la sangre (18,207).
PROMEDIO DE DATOS. Las variaciones de VE, ventilación-a-ventilación en ausencia de un aparente cambio en el estímulo, que puede ser considerado como «ruido», perturba las respuestas características. Sacando un promedio de las respuestas repetidas a un estímulo ventilatorio dado, alineándolas a marcadores codificados de los eventos, refuerza que la respuesta promedio se acerque más a la realidad y se aleje del «ruido». Los datos de experimentos sucesivos son colocados en orden para facilitar el cálculo del tiempo de la respuesta promedio y los índices de dispersión.
AJUSTE DE CURVAS. A veces se requiere un procesamiento de datos subsecuente. Se pueden promediar ventilaciones adyacentes para obtener una respuesta promedio a los estímulos en steady-state. En respuestas a estímulos progresivos, por ejemplo, es razonable ajustar líneas rectas con partes de los datos. Cuando relaciones exponenciales apropiadas pueden ser ajustadas para algunas variables en el comienzo del ejercicio, se pueden emplear algoritmos computados. También se pueden ajustar relaciones más complejas para datos ventilación-a-ventilación (ver ref. 249, y la sección siguiente).
B) Análisis de sistemas de la dinámica de tasas de esfuerzo forzadas.
Se han investigado los factores que contribuyen a las respuestas steady-state al ejercicio, para determinarlas respuestas características a dinámicas particulares de tasas de esfuerzo forzadas. Las teorías de sistemas de control ofrecen procedimientos matemáticos para el análisis de las respuestas de dinámicas forzadas de los sistemas fisiológicos (103,167). La perturbación de la tasa de esfuerzo puede ser considerada como el estímulo, y la respuesta ventilatoria como la respuesta (a pesar de que el VCO2, VO2, etc., también pueden ser considerados información de ingreso o respuesta, según se necesite).
IMPLICANCIAS DE LA LINEARIDAD. A veces es adecuado suponer que el sistema que relaciona el estímulo con la respuesta es lineal, o aproximadamente lineal. En la práctica, esto implica que si se amplía el estímulo de la tasa de esfuerzo, la forma de la onda de respuesta se ampliara simétricamente por el mismo factor. Además, si se aplican dos estímulos en forma simultánea, la respuesta es la suma de las respuesta a los estímulos individuales. La linearidad también implica que la derivada (o integral) de un estímulo dado, conlleva a la derivada (o integral) de la respuesta. Una consecuencia importante de la linearidad es que, si se conoce la respuesta hacia un estímulo, se puede predecir la respuesta a cualquier otro.
La relación entre tasa de esfuerzo y VE, aparentemente se puede tratar como cuasilineal. Durante el steady-state, la relación es una línea recta, al menos por debajo del umbral anaeróbico (UA)(235). Durante un ejercicio dinámico, si se evita la transición reposo-a-ejercicio, las condiciones de linearidad son satisfechas para algunas variables, por ejemplo el pulso, las etapas de esfuerzo y la carga generada por la inclinación (91).
Por lo tanto, las características dinámicas de la hiperpnea de ejercicio pueden ser estudiadas mediante varias formas de onda de tasas de trabajos forzadas. Se han reportado cinco formas de onda: escalonamiento (36,136,155,165), por pulso (11,91), en rampa (91,137), sinusoide (11,32,34,254,255), y secuencias binarias pseudorandomizadas (falso azar) (20). Sin embargo, no todas son igualmente ajustables a una aplicación dada. Los factores que hacen que una de estas formas sea más apropiada que otra son: 1) la cantidad de datos en respuesta obtenidos que es relevante para la dinámica; 2) la presencia de componentes de frecuencia apropiados en el ingreso; 3) la amplitud de la respuesta en relación al «ruido» ventilación-a-ventilación; 4) el tiempo requerido para la realización de un test de esfuerzo; y 5) el grado de interacción percibida por el sujeto con la tasa de esfuerzo.
DINAMICA DEL INTERCAMBIO GASEOSO. Los estudios recientes sobre la dinámica de respuesta de la hiperpnea en el ejercicio han ido más allá del solo hecho de examinar la VE, y se han extendido a variables asociadas, cuyas relaciones con la dinámica de la VE sirven para clarificar estas conexiones.
Las técnicas de computación para la medición ventilación-a-ventilación del VO2 y del VCO2 hicieron posible examinar, en detalle, la cinética del intercambio gaseoso (21,32,34,155,249). A pesar de que en el estado de equilibrio (steady-state), la cinética de este intercambio medida en la cavidad bucal refleja precisamente el intercambio gaseoso en los tejidos metabólicos (producción de CO2 y consumo de O2), dinámicamente esto no es verdad. Los cambios en la reserva gaseosa de tejidos y pulmones, tanto como las demoras en el transporte, disocian la cinética del intercambio gaseoso metabólico de aqué1 medido en la boca. Mucha de la información acerca de la hiperpnea del ejercicio ha sido extraída de la relación de la cinética de VE con la del intercambio gaseoso (ver COMPORTAMIENTO DEL SISTEMA DE CONTROL VENTILATORIO -en la siguiente página-, y DESARROLLO CONCEPTUAL DE LAS HIPOTESIS DE CONTROL VENTILATORIO -más adelante en el texto-). Sin embargo, una consideración clave es que el balance de masas dictamina que la cinética de VE sea un determinante de la cinética del intercambio gaseoso medido. Casaburi, Wasserman, Whipp, y cols. (33) han mostrado (en perro) que, a pesar de que la cinética del VO2 só1o está débilmente influenciada por marcados cambios en la cinética de VE al comienzo del ejercicio, la cinética del VCO2 es alterada dramáticamente.
Presumiblemente, esta distinción proviene de la acumulación corporal mucho mayor de CO2 que de O2 (38), y de la capacidad para producir un cambio en el contenido de CO2 arterial inducido por el VE; el contenido de O2 arterial no es sensitivo a los cambios en VE debido a la configuración plana de la curva de disociación de oxihemoglobina, a valores normales de PaO2 al nivel del mar. Por lo tanto, interpretar la relación de las cinéticas de VE y VCO2 depende en gran medida si VE se adelanta o se atrasa con respecto al VCO2, y en el cambio concomitante de PaCO2 (32).
EVALUACION DE LA HIPOTESIS. Los datos de los estudios de fuerza dinámica, en general, son ajustados a relaciones matemáticas para ayudar a la evaluación de la hipótesis. Como un primer paso, las expresiones matemáticas pueden ser ajustadas a los datos de la respuesta sin implicar que la forma de la expresión está relacionada con la estructura del sistema: la expresión es la más simple que se ajuste a los datos apropiadamente. Cuando las velocidades relativas de respuesta son de primordial interés, este enfoque puede ser útil (32,34,68).
Un segundo paso en la evaluación de la hipótesis demanda que la relación elegida esté estructuralmente relacionada con el sistema bajo condiciones de análisis dinámico, esto es que sea un modelo matemático. A pesar de que es claramente imposible identificar todos los procesos fisiológicos involucrados en la relación de la tasa de esfuerzo con la ventilación, se pueden probar las hipótesis acerca de la estructura del sistema determinando si un modelo describe los datos significativamente mejor que otro. Engeman y cols. (84) han demostrado que, si es requerida una discriminación entre dos modelos alternativos específicos, se puede elegir una forma de curva de ingreso de datos que optimice esta discriminación.
COMPORTAMIENTO DEL SISTEMA DE CONTROL VENTILATORIO
Respuesta ventilatoria al ejercicio progresivo
Aumentar la tasa de esfuerzo progresivamente desde un rango leve a moderado en una bicicleta o cinta ergométrica, aumenta la VE en forma lineal (Figura 1). Cuando la tasa de esfuerzo causa acidosis metabólica como resultado de los mecanismos anaeróbicos que soportan, en parte, la tasa metabólica [ej., una tasa por encima del umbral anaeróbico (UA); (170,235)], el incremento de VCO2 y VE se acelera mientras VO2 continua aumentando linealmente (53, 226, 230). Por varios minutos los incrementos acelerados de VCO2 y VE son paralelos, por lo que la PCO2 de final de espiración (PET CO 2) permanece relativamente constante.
El aumento acelerado de VCO2 en relación al VO2 es consecuencia del CO2 adicional generado por el tamponamiento (buffering) de ácido láctico por el HCO3 (39, 70, 226). Los incrementos paralelos iniciales de VE y VCO2 dan como resultado un período de «buffering isocápnico» (230) durante el cual los mecanismos de control parecen responder en proporción al cambio de CO2 generado, pero no responden a la disminución del pH. Debido a que el incremento de VE durante un test progresivo (ver Figura 1) se acelera, por encima del UA, mientras que el VO2 continua aumentando linealmente, la PO2 de final de espiración (PET O2) aumenta (ej., hiperventilación con respecto al O2) (225, 227, 235, 245). A medida que la tasa de esfuerzo se incrementa aún más, PET CO 2 disminuye (225, 227, 235, 245), reflejando una compensación respiratoria para la reducción del pH (acidosis metabólica). El período de «buffering isocápnico» se observa cuando los incrementos en la tasa de esfuerzo son durante períodos cortos, por ej. < a 2 minutos (Figura 1). Los tests en los cuales el tiempo entre cada incremento en la tasa de esfuerzo es relativamente largo (> o igual a 4 minutos), normalmente están asociados con una compensación respiratoria hacia el final de la primera tasa de esfuerzo por encima del UA (139, 226, 227, 235, 237). Davis, Whipp y Wasserman (57) han confirmado, en la actualidad, a partir de mediciones de gases sanguíneos arteriales y del pH que Pa CO2 permanece constante y que el pH disminuye, durante el período de «buffering isocápnico» detectado por mediciones de gases en los volúmenes de final de espiración (end-tidal) como aquellos valores de la Figura 1.
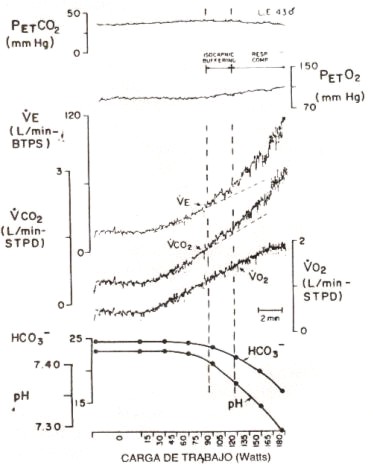
Figura 1. Medición ventilación-a-ventilación de la presión parcial de
final de espiración de CO2 y O2 (PET CO2 y PE
0), ventilación espiratoria por minuto (VE), producción de CO2 (VCO2),
consumo de O2 (VO2); en curvas graficadas por puntos, se
cuantifican las mediciones de HCO3 arterial y pH, durante el test
de cargas progresivas cada 1 minuto en bicicleta ergométrica en un sujeto
normal. «Buffering isocápnico», se percibe cuando VE y VCO2
aumentan exponencialmente en la misma proporción, manteniéndose la PET CO2
constante, con incremento de la PET O2. La «Compensación
respiratoria- (resp comp) representa el período cuando la PET CO2
comienza a disminuir. [Adaptado de Wasserman (225)].
Respuesta ventilatoria al ejercicio de carga constante
La respuesta ventilatoria al ejercicio de carga constante está caracterizada por tres fases distintivas (53, 64, 230, 244), tanto para el ejercicio como para la recuperación, como se esquematiza en la Figura 2.
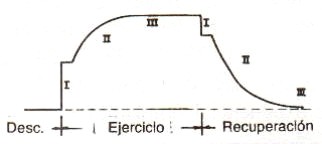
Figura 2. Esquema general de ventilación durante el ejercicio y la
recuperación. La fase I identifica el período de intercambio abrupto durante
la transición entre reposo y el ejercicio. La fase II identifica el período de
lento cambio hacia el steady-state (fase III).
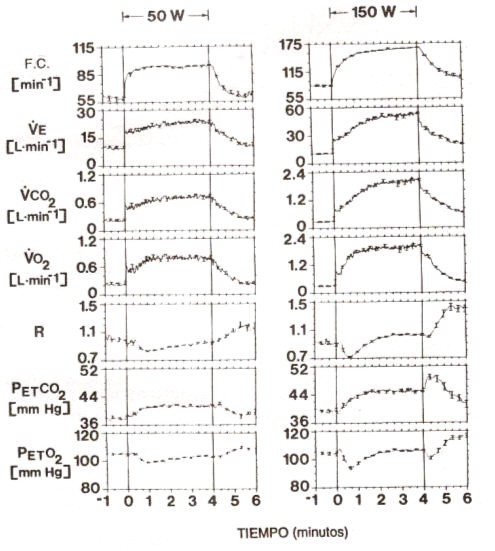
Figura 3. Medición continua de la ventilación e intercambio gaseoso de
un sujeto durante el ejercicio realizado a partir del estado de reposo, hacia
50 W y 150 W (media de estudios ventilación-a-ventilación hechos 8 veces a
cada tasa de esfuerzo). Las barras verticales,± 1 SEM, F. C.: frecuencia
cardíaca; R, cociente respiratorio. El sujeto comienza con la orden en minuto
0 y se le pide que se detenga al minuto 4. La velocidad de la bicicleta es de
60 rpm. cuando el sujeto comienza, por lo que no se agrega más carga, mas allá
de la necesaria para mantener el trabajo. La fracción de la respuesta
ventilatoria total atribuible a la fase 1 disminuye en relación a mayores
tasas de esfuerzo. Además, PET CO2, PET O2 y R no
cambian durante la transición del reposo al ejercicio. Notar la caída pasajera
de R y PET O2 que comienza a -20 seg.
La primer fase, al comienzo y al cesar el ejercicio, generalmente ocurre dentro de la primer ventilación [Figura 3; (56, 64, 127, 144, 179, 220, 247)]. La segunda fase ocurre luego de ~ 15 seg. y está bien descripta por una exponencial simple (28,37,65,68,155,253), con un tiempo constante de 60-70" (28,155,253). Dentro de ~ 4 minutos, VE alcanza un nivel constante (fase III) si el ejercicio está realizado por debajo del umbral anaeróbico del sujeto (Figura 4).
Por encima del UA, VE aumenta lentamente durante la fase III (155, 225, 237, 246, 253). Este lento incremento en VE para el ejercicio realizado, por encima del UA, está generalmente asociado con un incremento en la frecuencia respiratoria, mientras que VT es típicamente constante o puede disminuir levemente a medida que continua el ejercicio. Esto contrasta con la frecuencia fija y VT sobre períodos prolongados de esfuerzo realizado a un nivel inferior al UA (Figura 4). Debido a que las características de la ventilación difieren para las tasas de esfuerzo por debajo y por encima del UA, las mismas se consideran en forma separada.
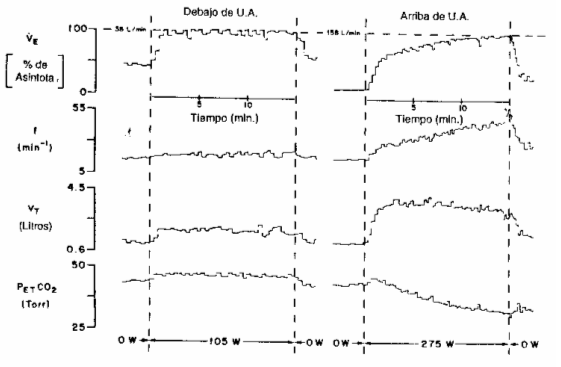
Figura 4. Cambios en VE, frecuencia respiratoria (f), volumen corriente
(VT), y PET CO2, durante un ejercicio en bicicleta ergométrica y
recuperación en un sujeto (promedio 8 ventilaciones). En el panel de la
izquierda, la performance con una tasa de esfuerzo por debajo del umbral
anaeróbico (AT). En el panel de la derecha, los resultados del mismo sujeto
por encima de su umbral anaeróbico. La tasa de esfuerzo comenzó, en la línea
de base, con pedaleo sin carga y se incrementó a los tiempos indicados.
POR DEBAJO DEL UMBRAL ANAEROBICO. Las variables de intercambio gaseoso y VE están graficadas, ventilación-a-ventilación, en la Figura 3. En sujetos normales, debido a la disminución en la capacidad funcional residual, típica en la primera ventilación luego del comienzo del ejercicio, VE puede ser transitoriamente mayor que la ventilación inspiratoria (127, 155, 222).
Simultáneamente la medición de VO2 y VCO2 revela cambios abruptos durante la fase 1, similares a aquellos de VE (Figura 3). Así, el cociente respiratorio gaseoso (R, o VCO2/VO2), PET CO2 y PET O2 no cambian durante los primeros 15 segundos, luego de la transición del reposo al ejercicio o del ejercicio a la recuperación (36, 127, 155, 220, 230, 242, 246, 253) (excepto para las respuestas «con sobresaltos» o cuando fisiólogos respiratorios entrenados actúan como sujetos). Por lo tanto, a pesar del incremento inmediato en VE, la sangre pulmonar, característicamente, no es ni hiperventilada ni hipoventilada (por ej. R no aumenta ni disminuye). De esta manera el volumen minuto cardíaco (Q; flujo sanguíneo pulmonar) debería haber aumentado tan abruptamente como el VE al comienzo del ejercicio, y disminuido tan abruptamente al final.
Debido a la mayor solubilidad del CO2 en los tejidos, en comparación con el O2, parte del CO2 metabólico es almacenado durante el ejercicio en condiciones de «no steady-state»,y no es intercambiado en los pulmones. Consecuentemente, R disminuye durante los siguientes 30 seg. antes de aumentar a su nivel de steady-state [Figura 3; (155, 180, 187, 230, 244, 253)]. Claramente, sin embargo, VE persigue a VCO2 durante la fase II (34,36,68,114,248,249); a causa que VE sigue a VCO2 más que a VO2 durante la fase II, PET O2 transitoriamente decae en respuesta al steady-state (Figura 3), causando una hipoventilación con respecto al O2 (155, 180, 234, 237, 253). Esto también ha sido
observado en muestras directas de sangre arterial (264). Cuando las
reservas de CO2 en el tejido se vuelven estables, VE alcanza un
steady-state y PaCO2 se vuelve constante. El incremento en PET CO2
durante la fase II (Figura 3) se debe a un aumento en la pendiente de la fase
alveolar de PCO2, a medida que aumentan el PCO2 venoso
mixto y Q; la PaCO2 media no cambia desde el nivel de reposo (244).
Las tensiones gaseosas y R permanecen sin cambios durante la recuperación a
través de la disminución del VE en la fase I (Figura 3). Durante la fase II de
la recuperación, R aumenta marcadamente y luego lentamente retorna a los
valores de reposo (155, 180, 187, 234, 237, 253) a medida que se exhala el CO2
acumulado en las reservas de los tejidos durante el ejercicio.
POR ENCIMA DEL UMBRAL ANAEROBICO. La fase I por encima del UA es similar a la observada por debajo del UA (Figura 3). Sin embargo, la fase II no termina claramente en un steady-state por encima del UA (155, 237, 246, 253), porque VE continua aumentando en respuesta a la acidosis durante la fase III (Figura 4). De esta manera, el precoz steady-state observado por debajo del UA se pierde, empañando la distinción entre la fase II y la fase III (Figura 4). Debido a que VE continua aumentando durante la fase III por encima del UA, PET CO2 disminuye y PET O2 aumenta (155, 236, 237, 246, 253).
Durante la recuperación las distintas fases son similares a aquellas por debajo del UA, excepto que VE permanece elevada por más tiempo a medida que el disturbio ácido-base que ocurrió durante el ejercicio es gradualmente corregido (ver próxima sección).
REGULACION DE LOS GASES SANGUINEOS ARTERIALES Y EL PH. Para las tasas de esfuerzo en el UA o menores (esfuerzos moderados), el pH arterial (pH a) y PaCO2 son regulados, cercanos al nivel de reposo (Figura 5). En forma similar, PaO2 permanece aproximadamente, a valores de reposo. Para los esfuerzos realizados por encima del UA, PaCO2 disminuye progresivamente (Figura 5B; también reflejado en la disminución de PET CO2 en la Figura 4). El pH a disminuye rápidamente y luego se nivela o vuelve a aumentar al valor de control (reposo), dependiendo de la severidad de la acidosis y del grado de compensación respiratoria (Figura 5C). Por lo tanto, pH a y PaCO2 no tienen los mismos patrones de cambio por encima del UA. El mecanismo de control continúa llevando a PaCO2 hacia abajo mientras restringe la disminución del pH a
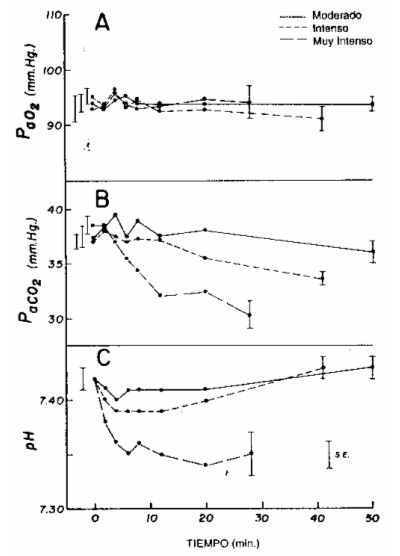
Figura 5. Efecto de un ejercicio prolongado a una tasa constante de
esfuerzo sobre las presiones parciales de O2 (PaO2) y CO2
(Pa CO.) en sangre arterial (A y B, respectivamente), y sobre el pH (C) para
un esfuerzo moderado, intenso, y muy Intenso. Cada punto representa la media
de 10 sujetos. (De Wasserman y cols. (226)).
Durante la recuperacion de un ejercicio muy intenso, el pHa es corregido mas rapidamente hacia los valores de control, mientras que la hiperventilaci6n persiste tanto tiempo como el HCO3 permanezca reducido, to que depende de la tasa de remoción de lactato (Figura 6). Asf, la prolongada hiperventilaci6n durante la recuperaci6n de un ejercicio muy Intenso está acoplada a la tasa de corrección de la acidosis metabólica del mismo.
El comportamiento de los mecanismos del control ventilatorio durante el ejercicio y la recuperación para los trabajos realizados por encima del UA parecen dirigidos a la homeostasis del pH a, aexpensas de la regulación de la PaCO2.
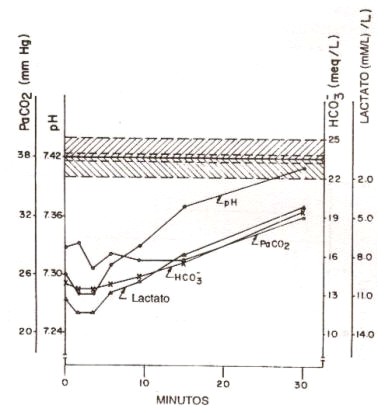
Figura 6. pH arterial, PCO2, HCO3- y lactato,
durante la recuperación de un esfuerzo muy intenso (el tiempo 0 es el valor
dentro del minuto antes de finalizar el ejercicio). Las líneas externas
horizontales de guiones, delimitan el rango de valores control para el pH
arterial, PCO2 y HCO3. Las líneas de guiones internas,
representan la media de la concentración de lactato sanguíneo de reposo. Cada
punto expresa la media de 4 sujetos físicamente estresados al mismo nivel. Las
curvas de cada sujeto conforman el promedio. Notar que el pH se corrige
primero. El lactato, PaCO2, y HCO3 se recobran
paralelamente.
Factores que afectan la respuesta ventilatoria al ejercicio de carga constante
FASE I. La magnitud de la fase I relativa a la respuesta ventilatoria en steady-state es la más grande a la tasa de esfuerzo más baja [Figura 3; (63, 126, 180)]. A medida que aumenta la tasa de esfuerzo, la proporción de la respuesta ventilatoria total que puede ser responsable del incremento inmediato del VE, es reducida. Por lo tanto, para el pedaleo sin carga, la fase I podría ser = ≥ 80 % de la respuesta ventilatoria total. Para una tasa de trabajo intensa puede ser < 25 %. La respiración de gases hipercápnicos (53,67,220), hipóxicos, o hiperóxicos (36,52) no parece afectar la magnitud de la fase I. Además, la fase I está presente en un grado normal en sujetos con una función pulmonar normal o cerca de lo normal, en quienes los cuerpos carótidos han sido quirúrgicamente resecados (236).
El incremento abrupto en VE en la fase I es sólo típico de las transiciones reposo-a-esfuerzo. Cuando el ejercicio es instituido a partir de una actividad previa suave, como el pedaleo sin resistencia, sólo se observa un pequeño componente temprano (20, 91, 155, 249), o no hay componente en la fase I, que es discerniblemente diferente del curso de tiempo de la respuesta de la fase 11 (5, 28, 36, 249).
FASE II. El tiempo medio, o la constante de tiempo (r), para la fase II de VE, VCO2, y VO2 ante un ejercicio moderado a carga constante (ya sea en reposo o con pedaleo sin resistencia) refleja un cercano acoplamiento dinámico de VE a VCO2, con una relación mucho más pobre de VE con VO2 (Figura 7). Las perturbaciones sinusoidales de la tasa de esfuerzo revelan las mismas relaciones (34). La fuerte correlación entre los valores de r para VE y VCO2 podrían no ser sorprendentes debido a que el CO2, siendo altamente soluble en la sangre, podría ser expelido como consecuencia de un incremento primario de VE. Sin embargo, varios investigadores han demostrado que la rVE es, actualmente, levemente más prolongada que NCO2 (34, 36, 68, 155, 249). Además, con una fuerza sinusoidal, la proporción VE/VCO2 es baja cuando VE y VCO2 son altos (34,36,252), y no a viceversa. Estas observaciones indican que el VCO2, en general, adelanta más que retrasa la respuesta ventilatoria, y que un pequeño error en flujo de descenso de PaCO2 y [H+] puede resultar durante la fase II (ello es apropiado en relación a los circuitos de «feedback» humoral). Ha sido documentado, con muestras directas de sangre arterial (252), un pequeño pero significativo incremento en PaCO2 cuando VE aumenta durante una tasa de esfuerzo que varía sinusoidalmente.
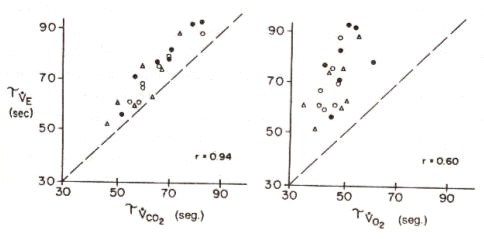
Figura 7. Comparación de las constantes de tiempo (r) para VE y VCO2
(panel izquierdo), y para VE y VO2 (panel derecho), desde el
comienzo de un ejercicio a tasa constante de esfuerzo desde: a) situación de
reposo (); b) transición de los 25 W hasta el umbral anaeróbico del sujeto
durante pedaleo a una tasa constante () ; y c) la misma transición de la tasa
de esfuerzo, desde los 25 W hasta el umbral anaeróbico del sujeto, pero con
aumento de la tasa de pedaleo de 40 a 80 r. p.m. () Todos los puntos están
por encima de la línea de identidad, indicando que las dinámicas ventilatorias
son más lentas que las de VCO2 y de VO2. Las dinámicas
ventilatorias están estrechamente correlacionadas con las de VCO2,
pero levemente relacionadas con las de VO2 [Adaptado de Diamond,
Casaburi, Wasserman y Whipp (68)].
El estrecho acoplamiento de VE a los cambios de VCO2 durante la fase II, sugiere que la ventilación podría estar bajo un control quimiorreceptor (con el CO2 y H+ como los posibles estímulos), y un quimiorreceptor regulador en el lado arterial. Por lo tanto, la cinética de la respuesta ventilatoria al ejercicio ha sido estudiada en pacientes asintomáticos a los cuales se les han extraído los cuerpos carótideos, pero cuya función era normal o estaba levemente dañada al momento del test. La respuesta ventilatoria al ejercicio de carga constante fue considerablemente más lenta que la de sujetos normales (236). También hubo una marcada elevación transitoria en la PaCO2 y PET CO2 antes de llegar a una respuesta en la fase III, en la cual PaCO2 fue indistinguible de los niveles de reposo (como los sujetos normales que practicaban ejercicios por debajo del UA). Estos resultados implican que los cuerpos carótideos son, al menos, parcialmente responsables por el estrecho acoplamiento de VE a VCO2 durante la fase II. Se llegó a la misma conclusión en estudios realizados por Casaburi, Whipp, Wasserman, y cols. (32), en los cuales los sujetos realizaron ejercicios en onda, durante los cuales respiraban aire o gas al 100 % de O2. Cuando la tasa de esfuerzo se variaba en una forma sinusoidal, la inspiración del gas al 100 % de O2 causaba que VE variara menos, y que PET CO2 variara más que con la inspiración de aire (Figura 8). Debido a que la inspiración de O2 atenúa la respuesta de los cuerpos carótideos en humanos, se puede concluir que estas estructuras son generalmente importantes para el acoplamiento de VE al VCO2 durante el ejercicio dinámico.
Además, Oren, Whipp y Wasserman (178) han mostrado que al inducir la acidosis metabólica crónica en humanos se estrechaba el acoplamiento de VE a VCO2, a pesar de que VE aún se retrasaba levemente al VCO2, mientras que la alcalosis metabólica parecía reducir el acoplamiento. Además, ellos observaron que respirar 100 % de O2 demoraba significativamente la respuesta ventilatoria de la fase II, al mismo nivel para los tres estados ácido-base. Los valores r para estas respuestas ventilatorias fueron similares a aquellos observados por Casaburi, Whipp, Wasserman y cols. (32), en condiciones de ejercicio sinusoidal hiperóxico (rVE -90 s). Consecuentemente, la acidosis metabólica acelera la dinámica de la respuesta ventilatoria por su acción sobre los cuerpos carótideos.
En estudios similares, Griffiths, Wasserman, Whipp y cols. (101) evaluaron la dinámica de VE durante la fase II en presencia de hipoxia e hiperoxia (Figura 9). El rVE fue considerablemente acortado por la hipoxia y prolongado con la hiperoxia. Por lo tanto, la diferencia entre rVE y rVCO2 es muy pequeña con la hipoxia o acidosis metabólica, y está aumentada con la hiperoxia o alcalosis metabólica.
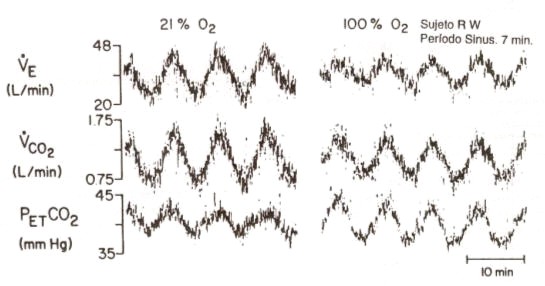
Figura 8. Mediciones de VE (BTPS), VCO, (STPD) y PET CO2 para
una tasa de esfuerzo que varía, en forma sinusoidal, entre 25 y 120 W
realizada por un sujeto normal respirando aire (panel izquierdo) y 100 % O2
(panel de la derecha). [De Casaburi, Whipp, Wasserman y cols. (32)].
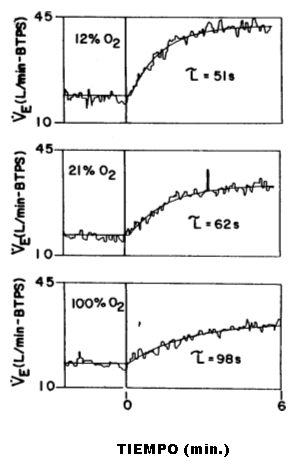
Figura 9. Respuesta ventilatoria a un ejercicio de 90 W a partir de una
línea de base de pedaleo sin resistencia o carga. Las dinámicas (constante de
tiempo) son claramente afectadas por la concentración de O2
inspirado.
Si la ventilación respondiera a factores mecánicos relacionados con la tasa de esfuerzo más que a factores humorales, entonces se podría esperar que VE, o un componente significativo de VE, variara en relación estrecha con el cambio real en la tasa de esfuerzo. Los estudios sobre el ejercicio con una onda sinusoidal realizados por Casaburi, Whipp, Wasserman y cols. (34,36) revelaron que a medida que aumentaba la frecuencia de la variación de la onda de la tasa de esfuerzo, el ángulo de la ventilación aumentaba en relación a la tasa de esfuerzo y la amplitud de la respuesta ventilatoria disminuía (Figura 10). Esto es característico de un sistema de control con un pequeño componente de respuesta ante la fase de fluctuación en la tasa de esfuerzo. Por lo tanto, para las ondas sinusoidales en períodos igual ≤ a 2', la ventilación varía marcadamente. Estos estudios sugieren que la ventilación no tiene un componente de control apreciable (< 10 %), que siga en forma cercana al stress inmediato en las extremidades que están en ejercicio, cuando la tasa de esfuerzo varía en forma sinusoidal. En cambio, VE durante esta forma de ejercicio mas parece responder en proporción al intercambio de CO2 pulmonar generado por el esfuerzo, con el mecanismo de control sin dar grandes respuestas a los variados cambios físicos en los músculos en ejercicio. Wigertz (254) ha llegado a observaciones cualitativamente similares.
Otras funciones de fuerza dinámica (pulso, step y fuerzas binarias pseudo randomizadas) han sido utilizadas para discernir los componentes rápidos de la respuesta ventilatoria (20, 84, 91, 92, 155, 249, 253). Ellas demostraron que la respuesta ventilatoria dinámica es consistente con un componente de 10-15 %, que es claramente no instantáneo pero parece tener una dinámica más rápida [r = 7 seg., estimado por Bennett y cols. (20), asumiendo una exponencialidad ] que el componente predominante (r = 70 seg.). Sin embargo, la interpretación mecánica de este pequeño componente debe considerar que un r de 7 seg. (por ej. requiriendo ~20 seg. para una respuesta del 95 %) es lento para un estímulo de ingreso neural de ]as extremidades en ejercicio, como está convencionalmente formulado. Además, Whipp, Wasserman y cols. (249,253) mostraron que aún en esta fase, la respuesta ventilatoria era proporcional al cambio de VCO2 y VO2.
Variar en forma sinusoidal la tasa de pedaleo durante el ejercicio a una tasa de esfuerzo constante no mostraba una relación entre la respuesta ventilatoria y el movimiento físico de las piernas (más allá de aquella causada por el leve costo metabólico del pedaleo a una mayor frecuencia) (35). Esta falta de asociación entre la respuesta ventilatoria y el ejercicio, con frecuencia de pedaleo en steady-state fue descripta por Craig y cols. (45), Sipple y Gilbert (199), además de Lloyd y Patrick (159). Por lo tanto, la tasa de movimiento de las extremidades no parece influenciar significativamente la respuesta ventilatoria al ejercicio.
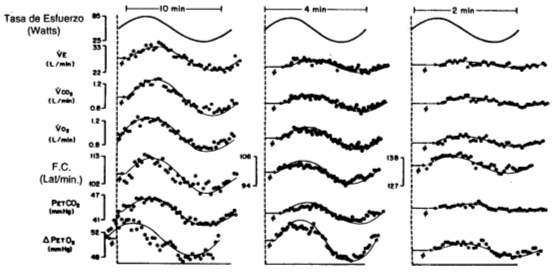
Figura 10. Amplitud y relación de las fases ( 0, retraso de la fase) para
una tasa de esfuerzo que varía en forma sinusoidal en períodos de 10, 4 y 2
minutos para un sujeto normal. Los puntos representan respuestas medias para
2, 6 y 10 ciclos sinusoidales, respectivamente. Las curvas son ondas
sinusoidales que mejor ajustan a los datos obtenidos. [de Casabud, Whipp,
Wassennan y cols. (34)].
FASE III. La respuesta ventilatoria al ejercicio durante la fase III se puede predecir menor a partir de la tasa metabólica (4, 52, 53, 72, 99, 102, 226). De esta manera, la respuesta ventilatoria durante el cuanto minuto (aproximadamente en steady-state) de ejercicio está relacionada en forma lineal con VO2 y VCO2 hasta elevados niveles de esfuerzo, cuando VE aumenta desproporcionalmente, en forma rápida [Figura 11, (226)]. Estos incrementos más rápidos en VE representan la compensación respiratoria para la acidosis metabólica producida por el ejercicio intenso (ver Figura 1), y parecen actuar predominantemente a través de los cuerpos carótideos (236,251). Hay una menor variabilidad entre los sujetos en el componente lineal de la respuesta ventilatoria, cuando este es relacionado con el VCO2 en comparación con el VO2 (226). La gran uniformidad de la respuesta es probablemente secundaria al estrecho rango en el cual la PaCO2 es regulada, y al patrón de cambio relativamente constante de la proporción: espacio muerto/ volumen corriente (VDS/VT), durante el ejercicio en sujetos normales.
La magnitud de la respuesta ventilatoria en el estado de equilibrio (steady-state) parece depender de la PaCO2 Oren, Wasserman, Davis y Whipp (177) demostraron con sales ácidas y alcalinas que las funciones en reposo de PaCO2 al incrementarse el CO2 a un punto determinado (CO2 set point), es un determinante de la respuesta ventilatoria al ejercicio (Figura 12). Por lo tanto, cuanto menor es ese punto determinado de CO2, mayor será la respuesta ventilatoria a un determinado nivel de ejercicio.
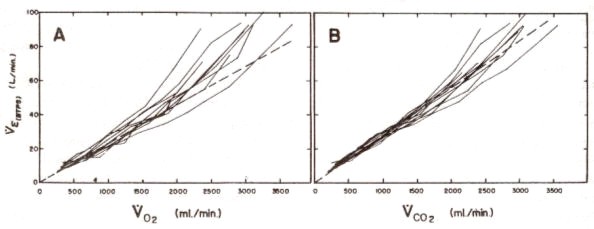
Figura 11. Relación de VE con VO2 (A) y con VCO2 (8)
en 10 sujetos normales. Cada curva es determinada, al menos, por seis
incrementos en la tasa de esfuerzo de 4 minutos de duración, con los valores
del cuarto minuto graficados [de Wasserman y cols. (226)].
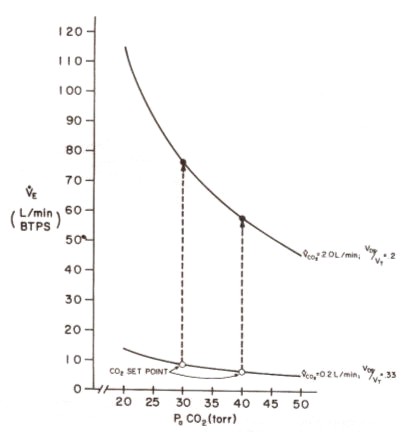
Figura 12. Curvas isometabólicas de VE computadas en relación a PaCO2.
La curva inferior, para VCO2 en reposo (0.21/min.) y tasa espacio
muerto / volumen corriente de 0.33 (VDSIVT). Curva superior, para VCO2
en ejercicio (2.0 1/min.) y VDSIVT (0.2). La longitud de las líneas de guiones
indican incremento en VE desde el reposo al ejercicio cuando se establece el
punto de PaCO2 (set point de CO) en 40 mmHg (derecha) y en 30 mmHg
(izquierda).
Wasserman, Whipp y cols. (236) compararon la respuesta en la fase III de sujetos con cuerpos carótideos resecados, con aquella de un grupo de sujetos normales, tanto por encima como por debajo del UA. En los sujetos sin los cuerpos carótideos la capacidad para regular la PaCO2 no fue perjudicada durante la fase III del ejercicio por debajo del UA, en comparación con el grupo control (236,251). Sin embargo, para el ejercicio por encima del UA, los sujetos sin cuerpos carótideos no pudieron desarrollar una compensación respiratoria para la acidosis metabólica y por lo tanto tuvieron una respuesta ventilatoria reducida (236,251).
La importancia de los cuerpos carótideos en la fase III del ejercicio también puede ser evaluada cambiando discretamente el gas inspirado, de aire a 100 % de O2. La magnitud del decremento resultante en el VE refleja la contribución de los cuerpos carótideos (61, 62, 200, 228, 234, 251). Para el ejercicio por debajo del UA esta maniobra no tiene un efecto sostenido discernible (Figura 13). Só1o provoca una pequeña (15 %) reducción transitoria de VE que dura aproximadamente 30 segundos (224). Por el contrario, para el ejercicio por encima del UA, la respuesta ventilatoria a esta maniobra es tanto rápida como sostenida con cambios concomitantes en PET CO2 (Figura 13). Esto apoya el concepto que los cuerpos carótideos brindan la compensación respiratoria para la acidosis metabólica aguda durante el ejercicio intenso.
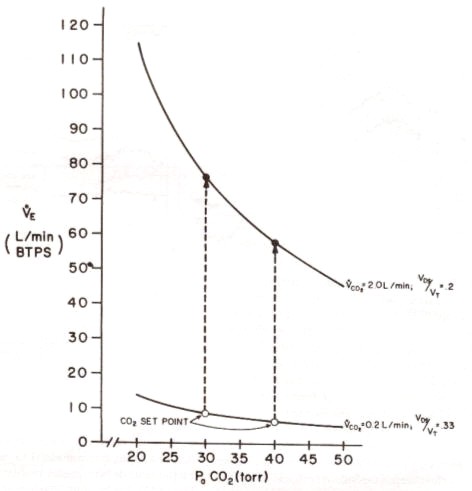
Figura 13. Efecto del cambio en la concentración de O2
inspirado sobre la ventilación para tasas de esfuerzo por sobre (panel
izquierdo) y por debajo (panel derecho) del umbral anaeróbico (UA) para un
sujeto. Panel superior, el gas inspirado fue cambiado de aire a O2
100 %; panel inferior, el gas fue cambiado de O2100 % a aire. Notar
el efecto del cambio sobre PET CO2 y VE [De Wasserman, Whipp,
Casaburi y cols. (231)].
Se ha sugerido que el flujo de CO2 al corazón y los pulmones está ligado causalmente a VE (157, 162, 185, 192, 229, 232, 233, 262). Debido a que el flujo de CO2 esta determinado por dos factores, Q y contenido de CO2 venoso mixto, VE fue medido mientras estos dos factores fueron modificados, de a uno por vez. De esta manera, Brown, Wasserman y Whipp (29) aplicaron propranolol (bloqueador Badrenérgico) en forma intravenosa, durante un periodo de 1 min., en sujetos normales que estaban en la fase III del ejercicio. La frecuencia cardíaca (y presumiblemente Q) disminuyó rápidamente, y VE y VCO2 siguieron con un cambio no significativo en PET CO2 A pesar de una disminución sostenida en la frecuencia cardíaca, VE retornó al nivel prepropranolol dentro de los 3 minutos, a medida que las reservas de CO2 y CO2 venoso mixto aumentaban, por ej., retornando el flujo de CO2 pulmonar al steady state previo al bloqueo. El bloqueo B- adrenérgico en sí mismo no tiene un efecto aparente sobre el control ventilatorio durante el ejercicio, más allá de sus efectos sobre el control cardiovascular [(182,204,214); ver también Relaciones Cardiovasculares Pág.611].
En forma similar, en perros anestesiados en ejercicio en los cuales VE y PaCO2 fueron constantemente medidos, Huszczuk, Whipp, Wasserman y cols. (124) desviaron parte del retorno venoso a la aorta inferior, luego que VE e intercambio gaseoso en el ejercicio habían alcanzado un steady-state (Figura 14). Inmediatamente a la desviación se produjo una hipopnea sostenida, mientras que PaCO2 en la aorta ascendente no cambia sistemáticamente. Luego de la desviación, VE retornó al nivel de ejercicio de control.
Figura 14. Efecto de la desviación parcial del retorno venoso desde la
aurícula derecha hasta la aorta inferior, durante el ejercicio, en dos perros
anestesiados. Las flechas negras que van hacia arriba indican cuando comenzó
la estimulación de los músculos con el propósito de inducir al ejercicio
muscular; las flechas negras hacia abajo, cuando se suprimió el estímulo. Las
flechas blancas indican cuando se aumentó el flujo del by-pass del
intercambiador de 0.51/min. a 31/min. y luego se disminuyó nuevamente a
0.51/min. EI perro A pesaba 40.5 kg; el perro B, 47 kg. Notar el cambio
abrupto en VE durante la desviación en A, con só1o una disminución transitoria
de PaCO, antes de retornar al valor de pre-desviación. También notar que en B,
PaCO, en realidad aumentó durante la desviación. Vexp: flujo de aire espirado;
PRV. presión ventricular derecha; BP: presión sanguínea arterial sistémica.
DESARROLLO CONCEPTUAL DE LAS HIPOTESIS DE CONTROL VENTILATORIO
El desarrollo conceptual del control de la hiperpnea en ejercicio no sigue una secuencia cronológica neta. Por lo tanto, nosotros hemos dividido esta sección de acuerdo a los avances conceptuales. Las hipótesis de control ventilatorio durante el ejercicio que han sido construidas sobre estos conceptos se muestran en la Figura 15.
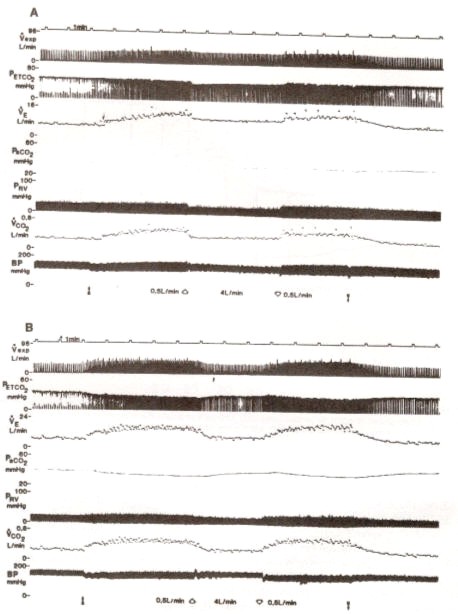
Figura 15. Hipótesis del control respiratorio durante el ejercicio.
VIA NEUROGENICA CENTRAL
VIA CORTICOGENICA. Las hipótesis responsables de la hiperpnea en ejercicio han sido dictadas en gran parte por el tipo de ejercicio utilizado en estos estudios. La mayoría de las teorías tratan de explicar el patrón de la respuesta ventilatoria al ejercicio muscular dinámico, con carga constante e intensidad moderada, el cual, como lo describió casi un siglo atrás Geppert y Zuntz (95), tiene respuestas tanto rápidas como lentas al steady-state. Los mecanismos de estas respuestas fueron formalmente estudiadas en 1913 por Krogh y Lindhard (144), quienes asumieron que la irradiación de signos nerviosos desde la corteza cerebral en ruta a los músculos en ejercicio, se desparramaba en el centro respiratorio en el cerebro, evocando cambios en VE proporcionales a la actividad muscular. Por lo tanto, ellos postularon que las respuestas rápidas (que ellos demostraron), que generaban una ventilación luego del comienzo del ejercicio eran mediadas neurológicamente, originadas en los centros más elevados. En su estudio, ellos no mencionaron las respuestas más lentas al steady-state. En seres humanos, solo se puede inferir una contribución cortical intrínseca a la hiperpnea en ejercicio, independiente de la hiperpnea volitiva. Por ejemplo, la hiperventilación ocurrida en el steady-state del ejercicio en situaciones, en donde el grado de esfuerzo consciente necesario para realizar la tarea era mayor que lo normal [luego de la parálisis muscular parcial por curarización (7, 176), por activación simultánea de la musculatura antagonista (97), o por sugestión hipnótica (55, 170)]. La sugestión hipnótica, diseñada para reducir el esfuerzo muscular aparente durante el ejercicio, disminuyó levemente la magnitud de la hiperpnea en ejercicio (55).
VIA HIPOTALAMICA. Eldridge y cols. (83) recientemente demostraron que estimular eléctricamente el área motora hipotalámica en el gato induce a rápidas respuestas ventilatorias y de intercambio gaseoso. Sin embargo, estos resultados no se parecen, cualitativamente, a aquellos obtenidos en estudios de ejercicio volitivo en seres humanos, o «ejercicio» inducido por estimulación de un músculo o nervio periférico en gatos o perros (146, 239, 240), o aun « locomoción ficticia» (83). O sea, la estimulación producía una profunda y rápida hiperventilación, y además, una abrupta hipertensión no característica del ejercicio. Esto, probablemente, resultaba de la estimulación de los circuitos extrapiramidales a los músculos respiratorios. Sin embargo, los experimentos nos hacen recordar que debemos considerar el rol del hipotálamo (y otras estructuras), en la respuesta integrada al ejercicio, de la misma manera que su rol en la inducción de la cólera, estado de alerta y defensa.
«REVERBERACION» NEUROLOGICA CENTRAL. Eldridge (79,81) también ha sugerido que los mecanismos neurológicos centrales pueden ser importantes en la determinación de la cinética de la fase H de la hiperpnea en ejercicio. Específicamente, el ha demostrado que los centros respiratorios en si mismos tienen dinámicas neurol6gicas que pueden sostener una hiperpnea a pesar de la remoci6n del estímulo causal. Más que una disminución inmediata en la actividad respiratoria, luego de que un cuerpo carótideo haya sido súbitamente extraído, o haberse suprimido un estímulo de esfuerzo en los miembros en acción, una caída temprana y abrupta de VE fue seguida por una disminución más lenta (79). El mecanismo de este componente más lento fue aislado del tronco cerebral. Un fenómeno similar también ha sido descripto en seres humanos en reposo, luego de que cesó súbitamente un esfuerzo de hiperventilación volitiva isocápnica (208, 211). Estos mecanismos pueden contribuir a la cinética de VE en la fase 11 del ejercicio, a pesar de que la simetría de comienzo y fin (on-off) del r en la fase H en humanos, no es tan evidente en los estudios de Eldridge en el gato (79, 81).
Vía neurogénica periférica
En 1932 en unas series de experimentos que mostraban una hermosa organización conceptual, Harrison y sus colegas (111), estudiando tanto al ser humano como al perro, concluyeron que la hiperpnea en ejercicio era controlada, en gran medida, por aferentes de las extremidades en ejercicio.
Sin embargo, esta teoría estaba en contra de las ideas de Winterstein (259), Haldane y Priestley (107,108), Douglas (70,71), Y Hasselbalch y Lundsgaard (112), quienes creían que la hiperpnea estaba en gran parte bajo control humoral, al notar el incremento proporcional de VE con la tasa metabólica.
A pesar de que varios investigadores subsiguientes han incorporado ambos conceptos, neurológico y humoral, en las teorías de hiperpnea en ejercicio, Dejours (60, 63, 64) ha brindado el análisis más sistemático y completo. El concluyó que, só1o una combinaci6n de mecanismos neurológicos y humorales podrían ser responsables del patrón de hiperpnea en ejercicio (Figura 2), con respuestas rápidas (fase 1) de origen neurológico, que ocurren muy luego del comienzo del ejercicio, y una lenta elevación subsiguiente al steady-state (fase II), la que es mediada en forma humoral. En el estado transitorio «off», ocurren respuestas direccionalmente opuestas pero cualitativamente similares. Esta teoría es conocida como teoría neurohumoral de la hiperpnea en ejercicio. Por lo tanto, nosotros consideramos la evidencia que apoya o contradice las ideas, que las respuestas rápidas al comienzo del ejercicio están mediadas neurológicamente, y que estas son sostenidas en steady-state mientras que los mecanismos humorales más lentos se agregan a ellas.
Varios investigadores (44, 111, 134, 166, 212) han seccionado o bloqueado los nervios aferentes de los músculos en ejercicio de perros o gatos anestesiados. Ellos demostraron que la ventilación aumentaba rápidamente al comienzo del esfuerzo pero que, cuando el flujo sanguíneo del músculo en ejercicio inervado era ocluido, la ventilación aún aumentaba más rápidamentee (44, 66, 111). Esto apoya la evidencia de un mecanismo neurológico en contra de uno humoral. Otros investigadores han estimulado realmente los nervios de los músculos en ejercicio; ellos han notado que se induce una hiperpnea. Por ejemplo, Bessou y cols. (22,23) demostraron que la estimulación máxima de los aferentes del grupo o tipo I de las extremidades aumentaba la ventilación, pero que las respuestas a la estimulación de los aferentes de los grupos II y III eran mucho más efectivas.
En forma similar, Senapati (196), Koizumi y cols. (141), Kalia y cols. (133), demostraron que la estimulación nerviosa de los músculos en ejercicio, especialmente de los aferentes más pequeños de los grupos III y IV, estimulaban la ventilación. Adicionalmente, sin embargo, Senapati (196), y Koizumi y cols. (141), observaron que los efectos de la estimulación duraban algún tiempo luego que el estímulo era removido.
AFERENTES DE LAS ARTICULACIONES Y MUSCULOS
La fuente de la vía aferente periférica ha sido tema de considerable debate. Comroe y Schmidt (44) creían que los receptores articulares estaban involucrados, punto de vista cuestionado subsiguientemente por Gardner y Jacobs (93). Sin embargo, Dejours y sus colaboradores (23, 60, 94, 150) creyeron que los husos musculares aferentes eran receptores más que importantes en el control. Gautier y cols. apoyan este concepto (94). Ellos inyectaron pequeñas dosis de succinilcolina en el flujo arterial al músculo de la pierna. Esta droga que estimula a los husos aferentes, inducía a una hiperpnea luego de unos pocos segundos. El cianuro, cuando se inyecta en el mismo sitio, demora 15 segundos en estimular la ventilación (hasta arribar a los quimiorreceptores periféricos). Estos investigadores pensaron que estos husos aferentes podrían conducir la ventilación a un nivel significativo. Flandoirs y cols. (89) realizaron el experimento anverso, inhibiendo la actividad del huso muscular farmacológicamente. Ellos demostraron que la inhibición reduce la magnitud de la respuesta ventilatoria al movimiento pasivo de las extremidades. Además, ellos demostraron que anestesiar los quimiorreceptores articulares de la rodilla con procaína reduce la ventilación.
Estos autores concluyeron que, en el perro, los mecanismos neurogénicos de origen en el huso muscular y articular eran importantes en el control ventilatorio. Por otro lado, Hodgson y Mathews (118) vibraron en forma longitudinal, el tendón del tríceps en el gato (un estímulo potente para los husos musculares) y concluyeron que los husos musculares aferentes parecen ser de poca importancia como un estímulo respiratorio. Sin embargo, ellos sugirieron que comprimir el músculo podría conducir a la hiperpnea. Pero Leitner y Dejours (150) afirmaron que, como Hodgson y Mathews solo habían estimulado un grupo del tríceps, la misma podría haber sido subliminal. Ellos repitieron los aspectos esenciales de este experimento, estimulando ambos grupos de tríceps y notaron que la ventilación aumentaba, aunque só1o en ~17 %.
Hornbein y cols. (122) infiltraron el espacio peridural lumbar con lidocaína para bloquear selectivamente el sistema γ-eferente en las extremidades inferiores de individuos en ejercicio.
Esto redujo, en gran medida, el reflejo de la rodilla pero no perjudicó, apreciablemente la fuerza muscular o la respuesta ventilatoria al ejercicio. Por lo tanto, ellos concluyeron que los husos musculares jugaban un papel mínimo en la hiperpnea del ejercicio.
Para identificar los tipos de fibras que acarrean la información aferente originada en la periferia, McCloskey y Mitchell (166) indujeron ejercicios en el gato por estimulación de la raíz ventral de la médula y registraron la actividad aferente proveniente de los miembros en las raíces dorsales. Ellos documentaron que la estimulación muscular rápidamente incrementa la ventilación. El bloqueo anodal de los aferentes del grupo I y II (incluyendo los aferentes de los husos musculares) no alteró significativamente esta respuesta ventilatoria. Sin embargo, cuando ellos indujeron un bloqueo anestésico de los aferentes de los grupos III y IV, el aumento ventilatorio fue suprimido en gran medida.
Estos resultados también apoyan aquéllos de Kao y colegas, de una serie de experimentos en el perro, los que se resumen en la referencia 134. Al impedir la información neurológica aferente de las extremidades en ejercicio con una cordotomía total, suprimió el incremento ventilatorio a la estimulación eléctrica del músculo en el perro neurológicamente aislado (la sangre de las extremidades en ejercicio fue circulada, en forma cruzada, a las extremidades de un perro en reposo, y viceversa). Kao también seccionó las columnas dorsales de la medula y observó que VE era reducido sólo levemente, mientras que cuando las columnas laterales eran seccionadas, la hiperpnea en ejercicio era suprimida.
Por lo tanto, los experimentos de Kao (134), McCloskey y Mitchell (166), Bessou y cols. (22,23), Kalia y cols. (133), y Senapati (196) apoyan los conceptos de que la estimulación de los aferentes en los grupos III y IV (pero no con la misma extensión en los grupos I y II), pueden estimular apreciablemente la ventilación. Aun se carece de una demostración crucial; los estudios no muestran que estos aferentes son estimulados durante el ejercicio de intensidad moderada. Por ejemplo, McCloskey y Mitchell (166) usaron contracciones tetánicas en sus experimentos, lo que podría estimular fibras dolorosas o causar acumulación de metabolitos capaces de estimular los aferentes no reclutados, normalmente, durante el ejercicio.
Más recientemente, sin embargo, varios grupos de investigadores (48, 152, 239, 240) han mostrado que, en animales experimentales con una sección del cordón espinal, o seccionando los nervios de las extremidades en ejercicio, o por sección de la raíz dorsal, no impedía los requerimientos de intercambio gaseoso de la hiperpnea en ejercicio (48, 240, 252).
En los seres humanos en reposo, la hiperventilación por varios minutos para vaciar las reservas de CO2 puede evitar que VE aumente al comienzo del ejercicio (5, 144, 223). No pasaran -30 segundos que haya continuado el ejercicio, cuando PET CO2 ya es reestablecida a los niveles de control, dado que VE se incrementa rápidamente hasta alcanzar la respuesta normal al ejercicio (223).
Este estudio sugiere que, si la neurogénesis es la base del incremento inmediato de VE al comienzo del ejercicio, la alcalosis respiratoria inducida puede fácilmente inhibirlo.
Adams y cols. (1,2) produjeron contracciones musculares de la extremidad inferior por estimulación eléctrica en parapléjicos que tenían una completa pérdida motora voluntaria y sensorial. Ellos observaron que VE aumentaba en proporción al incremento en VCO2 en el parapléjico, tanto como en los sujetos normales. Por lo tanto, los esfuerzos por documentar al estímulo neurogénico periférico como una causa importante de la hiperpnea en ejercicio no han sido uniformemente satisfactorios. Quizás, parte de la dificultad está relacionada con los marcados cambios en la circulación, metabolismo, y respuesta muscular consecuente al traumatismo agudo que genera la sección de la médula. Tales estímulos, luego de dicha sección, podrían alterar la contracción y la respuesta metabólica, y eliminar la estimulación de los receptores no relacionados con el ejercicio.
RECEPTORES METABOLICOS
La teoría que los tejidos periféricos podrían tener una función quimiorreceptiva estimulando la ventilación en proporción al cambio de la tasa metabólica del tejido, y ser mediados por aferentes neurológicos de las extremidades, fue propuesta en primer termino por Volkman (218) en 1841. Ha resurgido recientemente en el trabajo de Ramsay (188,189), Iaria y cols. (125), Kao (134), Liang y Hood (154), Levine (152), y Tibes y cols. (212). Kao y cols. (125,134) originaron el termino «ergorreceptor» para reflejar un receptor funcional del tejido que responde en proporción a la consecuencia del trabajo (tasa metabólica). Sin embargo, tales teorías, a pesar de ser al comienzo intuitivamente atractivas, carecen de evidencia que las apoye. A continuación siguen cinco ejemplos: 1) Varios grupos han introducido sangre conteniendo elementos de desecho metabólico de los tejidos periféricos, en el músculo, vía su suministro arterial (44, 94, 115, 134, 169), incluyendo sangre del efluente venoso de un músculo contráctil, y observaron que esto no induce a una hiperpnea apreciable. 2) La hiperpnea resultante por las sales de potasio en alta (pero no baja) concentración (158) estimula la ventilación pero, probablemente, también estimule los receptores no específicos de dolor. 3) Las condiciones que probablemente incrementan la hipoxia en el tejido periférico, tal como el ejercicio con inhalación de CO, que reducirían la PO2 venosa y del tejido, sin alterar PaO2, no aumentan la hiperpnea del ejercicio (6).4) La droga propranolol que reduce Q, bloqueando los receptores B-adrenérgicos, cuando se infunde durante el steady-state del ejercicio pareciera aumentar, transitoriamente, la acumulación de los productos del metabolismo en el músculo en ejercicio. Si los receptores metabólicos estuvieran presentes, VE se elevaría.
Sin embargo, Brown, Wasserman, y Whipp (29) han mostrado que VE cae transitoriamente, aparentemente en respuesta a una disminución pasajera del flujo de CO2 al pulmón. Esto argumenta en contra de una significativa fuerza, desde el tejido periférico, que genere la hiperpnea en ejercicio. 5) Tal interrupción de la información neurológica aferente de las extremidades falla en atenuar la respuesta ventilatoria, en los estudios de Adams y cols. (1,2), Cross y cols. (48), Lamb (146), Levine (152); y Weissman, Whipp, Huntsman y Wasserman (240), que también contradicen un importante rol de los mecanismos ergo receptivos o metabolo receptivos en la hiperpnea en ejercicio. Sin embargo, los resultados de Kao (134) y Tibes (212) no apoyan tal mecanismo. La base de estas observaciones, aparentemente contradictorias, probablemente esta en las diferencias en los sitios precisos y naturaleza de la deaferentación, y también en el estado anestésico de los animales.
Vía neurohumoral
Basado en el patrón de la respuesta ventilatoria al ejercicio en los seres humanos (rápido comienzo, luego lenta elevación hasta el steady-state, seguida por una disminución rápida y luego lenta del VE durante la recuperación), Dejours (64) postuló que la respuesta ventilatoria al ejercicio estaba compuesta por dos factores: 1) neurogénico (fase I, Figura 2), probablemente originado por los receptores mecánicos en las extremidades en ejercicio, y 2) humoral, comenzando 20-40 segundos luego del comienzo del ejercicio, y respondiendo a la acción de las catecolaminas, [H +], PCO2, y PO2 (fase II, Fig. 2), con una respuesta total (fase III), siendo ésta, la suma. Sin embargo, al profundizar esta teoría se observa que: l ) al comienzo del ejercicio, R no aumenta característicamente, y al final del ejercicio no disminuye [Fig. 3; (19,180)]; por ej., no hay ni hiper ni hipoventilación de la sangre en las transiciones del ejercicio; y 2) en los sitios de conocida quimiorrecepción no hay un incremento sistemático de [H +] y PCO2, y no hay disminución en PO2 que justifique el lento componente humoral del incremento en VE, al menos durante el ejercicio moderado.
Fue postulado que los cuerpos carótideos podrían ser responsables del componente humoral, operando con un incremento en la ganancia (51, 52) o respondiendo a cambios básicos en el pHa y en gases sanguíneos arteriales (26, 49, 50, 52, 54, 193). Sin embargo, un rol obligatorio de los cuerpos carótideos es inconsistente con la observación de que sujetos sin los mismos, o aquellos que respiran 100 % O2, tienen respuestas ventilatorias normales en steady-state de ejercicios moderados. Por lo tanto, atenuar la sensibilidad de los cuerpos carótideos no tiene un efecto depresor sostenido sobre VE en steady-state por debajo del UA (Figura 13). Además, es poco probable que las catecolaminas sean responsables del incremento del VE, ya que ellas son incrementadas principalmente durante el trabajo intenso (13, 60, 88, 106, 218a), y el bloqueo B-adrenérgico no afecta VE durante el ejercicio en steady state (29,182).
Conceptos humorales
Como se notó en la Figura 11 la respuesta ventilatoria en steady-state en sujetos normales (fase III) es muy uniforme, con una relación altamente predecible entre VE y VCO2. La precisión del mecanismo de control está reflejada en la regulación de PaCO2 en los valores de reposo, con los sujetos con un balance ácido-base normal. Durante el ejercicio de alta intensidad hay una compensación respiratoria por el desarrollo de la acidosis láctica (Figuras 1, 4). Estas observaciones sugieren que el estímulo a la hiperpnea en ejercicio puede ser totalmente (o en gran medida) humoral, con VE cambiando en proporción al flujo de CO2, para regular la composición del H + y el gas sanguíneo arterial. La búsqueda de los mecanismos para la regulación precisa de VE en la fase III, por debajo del UA, ha resultado en el desarrollo de varios conceptos, algunos apoyados por los datos experimentales.
Debido a que la fase lenta de la hiperpnea en ejercicio (fase II) comienza só1o luego de un tiempo proporcionado del transito de metabolitos de las extremidades en ejercicio hacia los quimiorreceptores (74), se sugirió que la fase lenta está bajo control humoral. En primer lugar se discute la fase III, en la cual el control humoral intenta alcanzar su máximo, y luego se considera la evidencia obtenida con respecto al control humoral durante la fase que no está en steady-state (fase II).
CONTROL «SIN ERRORES» EN EL STEADY STATE POR DEBAJO DEL UMBRAL ANAEROBICO. Cunningham (51) y Matell (165) han afirmado que gran parte del incremento en el steady-state en la hiperpnea en ejercicio puede ser generado por los aumentos en PaCO2 y [H +] arterial. Una considerable evidencia documentando que PaCO2 no aumenta sistemáticamente durante el ejercicio dinámico moderado, siendo regulada en su nivel de control, contradice esta teoría de control humoral (8, 77, 86, 110, 119, 120, 128, 147, 161, 164, 19, 194, 205, 226, 250). En aquellos casos en los cuales PaCO2 aumenta, esto se refleja típicamente en los muchos sujetos que hiperventilan al respirar inicialmente en una boquilla. Luego de que ellos entran en el steady-state y pierden este patrón hiperventilatorio de comportamiento, PaCO2 aumenta. Sin embargo, el incremento en PaCO2 generalmente retorna al nivel normal y no por encima de éste. En forma similar, la evidencia indica que pHa no cambia sistemáticamente en el steady-state de ejercicios moderados (Figura 5C).
Como consecuencia de la regulación de PaCO2 y pHa en sus niveles de control, y debido a que PaO2 no cambia sistemáticamente (Figura 5), han avanzado varias teorías para explicar el control «aparentemente sin errores», por ej., la hiperpnea isocápnica en steady-state. Defares (59), por ejemplo, ha sugerido que el «set point» para PaCO2 disminuye en proporción a la tasa metabólica. Grodins (102) y más tarde Swanson (206) recalcaron que un sistema de control incorporando un mecanismo de pro-alimentación (feedforward), ligado a la tasa metabólica, acoplado con un mecanismo humoral de control de retroalimentación, podría inducir respuestas ventilatorias típicas del ejercicio. Guenard y cols. (105) sugirieron un incremento en la sensibilidad o ganancia de la respuesta del CO2 (un concepto desafiado por Cunningham, quien notó la similitud en la pendiente de la curva de la respuesta del CO2 en reposo y durante ejercicio moderado). Grodins y James (104) y Wasserman, Whipp, Casaburi y cols. (231) sugirieron que el controlador puede operar como un integrador. Wasserman, Whipp, Casaburi y Beaver (229) también han hipotetizado que el sistema de control responde de algún modo a una sepal proporcional al cambio del flujo de CO2 a los pulmones durante el ejercicio. Esto tiene mucho en común con la brillante especulación de Yamamoto (261), y Yamamoto y Edwards (262), pero sin especificar que la mediación resulta de las características de la oscilación de PaCO2.
El rol del flujo de CO2 a los pulmones en el control de VE ha sido estudiado en animales experimentales, agregando o extrayendo CO2 del retorno venoso con un intercambiador gaseoso, a través del cual la sangre arterial o venosa es dirigida antes de que retorne a la vena central. En reposo o durante el ejercicio, VE disminuye cuando CO2 es reducido en el retorno venoso, y aumenta cuando CO2 es agregado al mismo. Sin embargo, desafortunadamente los informes no son consistentes sobre si el cambio en VE ocurre con poco cambio en PaCO2 (175, 184, 201, 217, 232, 252), o si la respuesta ventilatoria depende de un cambio en PaCO2, el que es definido por la curva de la respuesta del CO2 inhalado (101, 153, 185, 198).
Las diferencias en las observaciones obtenidas con técnicas de llenado y vaciado de CO2, probablemente pueden ser responsables de los efectos secundarios del procedimiento experimental, por ej., cambios en el retorno venoso (Q) (175, 185, 202), nivel de PaO2 (184), volumen sanguíneo central, diferencias en las especies, y anestesia.
Alterar el flujo de CO2 a los pulmones en una forma isocápnica afecta VE en seres humanos despiertos que no están realizando ejercicio. Dolan, Whipp, Wasserman y cols. (69) estudiaron los efectos ventilatorios de la alteración del contenido de CO2 venoso mixto en pacientes con una rutina de hemodiálisis. Con acetato como el principal buffer en la solución de diálisis, ~ 3 meq de HCO3 fueron extraídos del retorno venoso cada minuto (-60 ml de CO2/min.).
Esto afectó solamente el flujo de CO2 venoso porque el HCO3 era regenerado dentro de un tiempo de circulación, evitando una disminución neta de HCO3 arterial. Sin cambiar PaCO2 en forma significativa, VE disminuyó cerca de 1/3, en relación al decremento en VE predicho por la tasa de remoción de CO2 (HCO3) por la diálisis. Por el contrario, esta hipopnea no se presentó cuando se realizó la diálisis con los mismos pacientes con HCO3 como buffer en el líquido dializador. Estos resultados brindan la evidencia que el flujo de CO2 a los pulmones (por mecanismos que aun deben ser discernidos) es un determinante importante en la regulación ventilatoria.
Por otro lado, Foster y Crandall (90), Filley y Heineken (87), y Hill y cols. (117), realizaron un estímulo de CO2 en la sangre mediante perfusión de los quimiorreceptores, generando cambios que podrían no ser evidentes en la sangre subsiguientemente extraída y analizada. Estos investigadores sugirieron que, si la anhidrasa carbónica es restringida en gran medida al interior de los glóbulos rojos y es excluida del plasma (excepto durante la hemólisis), el H + no tenía un equilibrio, durante el transito, una vez superadas las superficies de intercambio gaseoso en el lecho capilar pulmonar. Por lo tanto, la reacción de hidratación podría continuar en las venas pulmonares y arterias sistémicas. Esto resultaría en un estímulo acidótico, que desaparecería cuando se alcance el equilibrio 1 minuto más tarde. Las muestras sanguíneas extraídas, convencionalmente analizadas, revelan este valor de equilibrio; los quimiorreceptores, sin embargo, sentirían y responderían a un pH más bajo. Esta teoría es atractiva en cuanto que la magnitud del estímulo de H + en la sangre, detectada por los quimiorreceptores, sería proporcional tanto para el CO2 venoso mixto como para el flujo sanguíneo pulmonar. Sin embargo, el concepto sufrió una contrariedad aparentemente irrecuperable, cuando Effros y cols. (75,76), Klocke (140), y subsiguientemente Crandall y O'Brasky (46), demostraron una fuente accesible de anhidrasa carbónica en el endotelio capilar pulmonar, permitiendo que el equilibrio de CO2 llegue a un virtual final durante el transito a través del pulmón.
De acuerdo a una tercer teoría, CO2 y H + estimulan la respiración a pesar de que PaCO2 y [H +] no cambian durante el ejercicio moderado (260-262). Debido a que la ventilación es una maniobra fásica, las oscilaciones de PCO2 y por lo tanto [H +] son impuestas sobre la sangre que abandona los capilares pulmonares (12, 98, 174, 186, 263). La amplitud de las oscilaciones de PCO2 y [H +] refleja el patrón de respiración y la concentración venosa mixta; la tasa de cambio instantánea de la oscilación de PCO2 refleja el gradiente instantáneo de PCO2 de la arteria pulmonar al gas alveolar. Una sepal proporcional a la tasa máxima de cambio de esta oscilación o a la amplitud pico fue sugerida como un mecanismo plausible para la hiperpnea en ejercicio. Esto también brindaría un estímulo potencial proporcional al flujo de CO2 en el retorno venoso. Varios grupos de investigadores creen que una fuente significativa del camino hiperpneico proviene de la transducción del cuerpo carótideo de tales señales oscilatorias (22, 49, 54, 262). Sin embargo, dos demostraciones eliminan tal mecanismo: 1) sujetos normales respirando 100 % de O2 y sujetos que no tienen cuerpos carótideos tienen la misma hiperpnea en steady-state de un ejercicio moderado que los sujetos control, que respiran aire (236); y 2) las oscilaciones de las tensiones gaseosas sanguíneas arteriales a diferentes frecuencias respiratorias serían mediadas por quimiorreceptores periféricos y no por mecanismos quimiosensitivos, centrales (163, 221).
Desde entonces se han sugerido dos modificaciones a la hipótesis de la oscilación. En primer término, Cunningham (54) hipotetizó que la importancia de la sepal oscilatoria a medida que es transducida por los quimiorreceptores, puede variar con la fase del ciclo respiratorio en la cual es detectada. Esta teoría está basada en la demostración de Black y Torrance (27), Dutton y cols. (73), y Eldridge (78,80), en las que un estímulo a los quimiorreceptores presentado durante la fase inspiratoria del ciclo respiratorio causa una estimulación ventilatoria, mientras que se evidencia muy poca estimulación cuando la misma alcanza a los quimiorreceptores durante la fase espiratoria.
Consecuentemente, un cambio de la fase de la sepal oscilatoria en una fase más sensitiva del ciclo respiratorio que se está llevando a cabo, podría brindar un estímulo hiperpneico. Este mecanismo ha sido denominado «acoplamiento de la fase» (244). En el steady-state del ejercicio en los seres humanos, el acoplamiento de la fase a las oscilaciones espontáneas de la tensión gaseosa sanguínea de [H +] parece no ser una fuente significativa de estimulación.
Petersen, Whipp y cols. (183) estimaron la fase del ciclo respiratorio donde el pico y las tasas máximas de cambio de [H +] arterial y PaCO2' era probable que ocurrieran en los cuerpos carótideos. Durante el steady-state de ejercicio moderado, no se pudieron encontrar diferencias significativas en el tamaño de aquellas respiraciones en las cuales los estímulos supuestos llegaban en la fase inspiratoria versus espiratoria de la respiración.
Biscoe (24), a pesar de que no expresamente en el ejercicio, ha sugerido una leve modificación de la hipótesis de acoplamiento de fase, en la cual el retraso de la fase entre la sepal oscilatoria que alcanza los cuerpos aórticos y carótideos, estimularía la respiración. Sin embargo, esta teoría es difícil de apoyar en los seres humanos ya que los cuerpos aórticos parecen no servir virtualmente para una función quimiorreceptora de control ventilatorio (121, 161, 209, 219, 251). Además, la ventilación parece ser normal en el steady-state de ejercicio moderado, en sujetos normales sin cuerpos carótideos. Saunders (193) ha propuesto que los cuerpos carótideos pueden realmente sentir el número de oscilaciones que pasan por ellos por unidad de tiempo, más que sentir la magnitud o máxima tasa de cambio de la oscilación. La misma crítica puede ser aplicada a este mecanismo propuesto.
QUIMIORRECEPTORES VENOSOS MIXTOS. Riley (191) y Armstrong y cols. (3) estudiaron quimiorreceptores venosos mixtos (o de la arteria pulmonar) que podrían estimular la ventilación (un sistema de alimentación adelantada). Sin embargo, muchos investigadores (10, 42, 43, 47, 58, 96, 116, 210) han explorado la posibilidad de la quimiorrecepción venosa mixta y no encontraron evidencia para tal mecanismo.
Todos los cambos en la ventilación podrían ser responsables por la estimulación de los quimiorreceptores periféricos o centrales. Sin embargo, Sheldon y Green (197), recientemente, reportaron estimulación de CO2 de la ventilación en un experimento con un perro en la cual se le realizaba, en forma separada, una perfusión en la circulación pulmonar aislada con sangre de varios niveles de PCO2. Estas respuestas eran inhibidas por una sección vagal; la relación de esta respuesta ventilatoria con el efecto del CO2 sobre los receptores pulmonares, aun no ha sido resuelta.
QUIMIORRECEPTORES PERIFERICOS. La despareja cinética de VE y de VCO2 en la fase II lleva a la predicción que la PaCO2 y [H + j arterial pueden incrementarse transitoriamente, y de este modo estimular los quimiorreceptores periféricos (predominantemente los cuerpos carótideos). Las observaciones que apoyan este mecanismo son un aumento en PaCO2 en la fase II (252) y una dinámica más lenta de incremento de VE en la ausencia de cuerpos carótideos (236), o durante la respiración con 100 % de O2 (Figuras 9, 13). A pesar de que el incremento de VE en la fase I, al comienzo del ejercicio, no depende de la presencia de quimiorreceptores conocidos (236), la respuesta ventilatoria durante la fase III en la ausencia de cuerpos carótideos (213,236), o durante 100 % de respiración de O2 (8, 14, 52, 60, 132, 241, 257) es reducida, pero só1o por encima del UA (Figura 13). Bajo tales circunstancias el componente de VE de la hiperpnea en ejercicio que provee una compensación respiratoria para la acidosis metabólica, está ausente o marcadamente atenuado.
QUIMIORRECEPTORES CENTRALES. Los quimiorreceptores centrales no parecen ser importantes como quimiorreceptores de control proporcional para la hiperpnea del ejercicio moderado. En estudios con animales, el fluido cerebroespinal no se vuelve acidótico durante una tasa de esfuerzo moderada (135,151) y puede, en realidad, volverse alcalino durante el esfuerzo intenso (25) a medida que el animal hiperventila.
ESTIMULOS RESPIRATORIOS NO RECONOCIDOS. Muchos investigadores en búsqueda de un estímulo humoral a VE en ejercicio, han postulado que una sustancia no conocida, liberada en el efluente venoso desde el músculo durante el ejercicio, aumenta VE (ref. 113). Más recientemente, esto ha recibido apoyo de estudios realizados por Levine (152). Sin embargo, aún no se ha encontrado tal agente en la sangre de perros en ejercicio.
Conexiones cardiovasculares
Un punto de vista completamente diferente del control del componente rápido de la hiperpnea en ejercicio ha sido expuesto por Wasserman, Whipp, Casaburi y cols. (229,233), quienes notaron que el rápido incremento en la ventilación al comienzo del ejercicio no esta fuera de proporción con el intercambio de CO2 en el pulmón (Figura 3). Ellos razonaron que, debido a que las tensiones gaseosas alveolares y el R no cambian en las primeras ventilaciones luego de la transición del reposo al ejercicio, o de un ejercicio suave a uno más intenso, la ventilación debe cambiar en proporción al cambio en el flujo sanguíneo pulmonar. Por lo tanto, o se debe invocar un estímulo paralelo, tanto a la ventilación como a Q, o se debe postular una sepal que estimule la ventilación en proporción a, y en respuesta, al cambio cardiovascular (Figura 15). Claramente, cualquier cambio en el flujo sanguíneo pulmonar que no es igualado por un aumento proporcional de la ventilación alveolar (VA) debe conducir aun error en PCO2, pH, y PO2, estímulos capaces de inducir hiperpnea a través de quimiorreceptores periféricos (u otros de respuesta rápida).
Para investigar tal mecanismo, Wasserman, Whipp y Castagna (233) estimularon Q con una pequeña cantidad intravenosa de hidroclorito de isoproterenol, o con estimulación eléctrica cardíaca directa; a medida que Q aumentaba, también lo hacía la ventilación, con poco o ningún cambio en PET CO2 Se concluyó que la respuesta ventilatoria era acoplada a un cambio en Q (hiperpnea cardiodinámica). La sugerencia de que la hiperpnea cardiodinámica operaba a través de un pequeño error transitorio en la tensión gaseosa, que resultaba en la estimulación humoral de los quimiorreceptores de respuesta rápida, ha sido desafiada por Winn y cols. (258), y Eldridge y Kumar (82). Estos investigadores presentaron evidencia de que los efectos ventilatorios de las inyecciones de isoproterenol eran el resultado de la estimulación directa de los cuerpos carótideos por la droga, más que un efecto indirecto de la droga mediado por el cambio en Q. Para resolver esta controversia, Juratsch, Whipp y cols. (131) aislaron temporalmente los quimiorreceptores conocidos del tórax interponiendo largas ligazones de retardo de flujo, tanto en las arterias carótidas como vertebrales (manteniendo cuidadosamente los flujos sanguíneos con bombas), en perros. La respuesta ventilatoria a la inyección intravenosa de isoproterenol tuvo dos fases. La primera pareció resultar del mecanismo cardiodinámico, y la segunda, de la estimulación directa de los quimiorreceptores. Esto evidenció que un mecanismo cardíaco podría incrementar VE en la fase I estimulando la ventilación con una sepal proporcional al flujo sanguíneo pulmonar.
Debido a la urgencia y magnitud de las respuestas ventilatorias iniciales, Whipp (243) cuestionó si los quimiorreceptores periféricos causaban hiperpnea cardiodinámica. La investigación fue basada en el tiempo de transito conocido desde los pulmones a los quimiorreceptores periféricos y la ganancia quimiorreceptora periférica. Wasserman, Whipp y cols. (236) brindaron mayor evidencia experimental, en la que los quimiorreceptores periféricos eran fuentes poco probables de la hiperpnea inicial, notando que la magnitud del incremento temprano de VE no difirió entre la gente con y sin cuerpos carótideos, y que la magnitud era similarmente no afectada por altas concentraciones de O2 inspirado, las que atenúan la quimiosensibilidad en los seres humanos, o bajas concentraciones de O2, las que aumentan la ganancia quimiorreceptora periférica (52, 53, 67, 173). Estas observaciones, por lo tanto, cuestionan el mecanismo propuesto de la hiperpnea cardiodinámica, pero no desafían su existencia.
Para evaluar con más profundidad la hipótesis de la hiperpnea cardiodinámica durante la fase I de la hiperpnea en ejercicio, Weiler-Ravell, Cooper, Whipp, y Wasserman (238) estudiaron las respuestas ventilatorias y de intercambio gaseoso en seres humanos ventilación-a-ventilación, desde el reposo al ejercicio en las posiciones vertical y supina. Este estudio tomó ventaja del hecho que el volumen sistólico en la posición supina ya es aumentado a aproximadamente aquel logrado durante el ejercicio en posición vertical, y por lo tanto solo sufriría un cambio menor al comienzo del ejercicio. Por lo tanto, el Q en reposo sería mayor en la posición supina, pero el incremento en Q en respuesta al período de la fase I del ejercicio sería menor, en comparación con la posición vertical. Ellos observaron que el aumento de VE en cada sujeto, durante la fase 1, era reducido en la posición supina, volviéndose una pequeña fracción de la respuesta total. Usando el incremento en VO2 para medir el aumento de Q durante la fase I (el contenido de O2 venoso mixto es casi igual al valor de reposo; por lo tanto, VO2 es proporcional a Q), ellos observaron que el incremento de VE en la fase I estaba linealmente correlacionado con el aumento de Q. Este estudio apoya el concepto que una respuesta modificada de Q al ejercicio también modifica la respuesta ventilatoria. Los estudios de Loeppky y cols. (160), y Miyamoto y cols. (168) también apoyan una conexión cardíaca al incremento de VE durante la fase I.
MECANISMOS DE ACCION DE ALIMENTACION ADELANTADA. Una nueva posibilidad para la mediación de la conexión de VE con Q ha sido sugerida por los estudios de Jones, Huszczuk y Wasserman (130), quienes demostraron quo cambiar el llenado ventricular derecho podría inducir a una hiperpnea significativa con una alta correlación entre estas variables (r = 0,9). Por lo tanto, estos investigadores sugieren que el mecanismo de hiperpnea cardiodinámica puede ser reflejo, originado en los receptores en la pared ventricular derecha. Los estudios electrofisiológicos han mostrado receptores en el ventrículo derecho con aferentes en los nervios vago (40) y simpático (143, 215). Se han documentado (142, 216) los efectos reflejos de la distensión ventricular derecha sobre la respiración. Se han descripto reflejos similares originados en la arteria pulmonar y que viajan en aferentes vagales (149). Los estímulos de la aurícula derecha también han sido postulados como una conexión a la ventilación (216).
Este posible mecanismo esta en una categoría diferente del original sugerido como mediador de la hiperpnea cardiodinámica. El concepto original sugería un mecanismo de retroalimentaci6n que respondía a un error humoral transitorio, consecuente a un incremento de la perfusión fuera de proporción con el incremento ventilatorio. Sin embargo, los cambios en la respiración causados por cambio en el llenado ventricular derecho o retorno venoso alterado sugieren quo el mecanismo es de alimentación adelantada, conduciendo la ventilación en proporción al llenado ventricular derecho (y por lo tanto Q). La regulación de la tensión gaseosa sanguínea indicaría que la sepal ventricular derecha al centro respiratorio induce a una hiperpnea en proporción al incremento del flujo sanguíneo pulmonar. Presumiblemente, los errores transitorios en las tensiones gaseosas y en el pH, quo ocurren por un desbalance en esta proporción, estimularían los quimiorreceptores y brindarían una corrección de retroalimentación de cualquier respuesta inadecuada de alimentación adelantada, como es sugerido por la respuesta de PaCO2 registrada directamente al cambio agudo en el retorno venoso mostrado en la Figura 14. Por lo tanto, la evidencia ahora indica que el cambio proporcional en VA y Q, al comienzo del ejercicio, no es una coincidencia pero podría ser un reflejo que acopla VE a Q, regulando las tensiones gaseosas sanguíneas al comienzo del ejercicio.
RETROALIMENTACION. A continuación se expone la evidencia que indica que la hiperpnea cardiodinámica también puede funcionar como un mecanismo de retroalimentación.
QUIMIORECEPTORES ARTERIALES. Un quimiorreceptor arterial sensitivo (por ej., cuerpos carótideos) podría detectar un error químico resultante de alteraciones de VA/Q. Esta hipótesis es apoyada por los resultados de los estudios cardíacos de Jones, Wasserman y cols. (129) en seres humanos en reposo con bloqueo cardíaco. Cuando se le ordenó el ritmo al corazón y se aumentó Q, VE aumentó, retrasando levemente el aumento en PET CO2. Cuando el ordenamiento fue discontinuo, VE disminuyó, nuevamente retrasando levemente la disminución de PET CO2. Los estudios con animales experimentales varían con respecto a la sensibilidad de los quimiorreceptores arteriales periféricos. Sin embargo, como los cuerpos carótideos parecen no tener umbral o acción en la pata del perro, para medir la respuesta neurológica al incremento en PCO2 (145), ellos son claramente sitios potenciales para detectar tales cambios. Se dispone de evidencia que demuestra que los cuerpos carótideos responden a cambios en el pH tan pequeños como 0,01 unidades (210).
RECEPTORES QUIMIOSENSITIVOS INTRAPULMONARES. En el perro, los receptores pulmonares vagales son estimulados por bajos niveles de CO2 intrapulmonar (16, 41, 123, 171, 195), disminuyendo VE (15,17). Concebiblemente en los seres humanos, debido a que a1gunas unidades pulmonares tienen una pobre perfusión, los receptores inhibitorios pueden ser estimulados de esta manera. Durante el aumento en el flujo sanguíneo pulmonar, al comienzo del ejercicio, las unidades pulmonares con una baja perfusión previa y con una baja PCO2, se transforman con una buena perfusión, disminuyendo de este modo la estimulación de los reflejos vagales sensitivos al CO2. Por lo tanto, las influencias inhibitorias del vago serían reducidas, y VE consecuentemente incrementada, en proporción con el flujo sanguíneo (y de CO2) pulmonar. Esta noción se asemeja al concepto de Hall (109), un fisiólogo inglés de mitad del siglo XIX de que la hiperpnea del ejercicio involucra un reflejo de CO2 intrapulmonar mediado vagalmente el cambio en las variables reguladas en respuesta al «stress». El mecanismo de control que aumenta la ventilación en respuesta al ejercicio es, normalmente casi ideal, porque responde prontamente al incremento en las tasas de intercambio gaseoso para mantener la homeostasis de PaCO2, pHa, y PaO2 durante el ejercicio moderado, y responde más rápidamente durante el ejercicio intenso para adaptarse al «stress» adicional de [H +].
Las descripciones de la respuesta ventilatoria al ejercicio en este capítulo derivan predominantemente de estudios con seres humanos. No todas las especies tienen precisamente las mismas características fisiológicas de control, y los estudios con especies no humanas pueden confundir nuestra búsqueda de mecanismos de control ventilatorio en los humanos. Por ejemplo, la regulación de la temperatura y [H +] podrían ser influencias que compiten en especies que dependen de la ventilación para controlar la temperatura corporal. Además, la interacción animal ser humano y las anestesias podrían ser influencias que se confunden. No obstante, los estudios con especies no humanas son útiles y necesarios para evaluar los mecanismos subyacentes. Sin embargo, será necesario retornar a estudios con seres humanos para determinar la importancia de cualquier mecanismo dado.
Se han descripto muchos mecanismos que podrían aumentar el VE bajo condiciones de ejercicio. Se necesita una mayor clarificación sobre cuales son las dominantes en brindar el acoplamiento, remarcablemente preciso, de la ventilación del ejercicio al intercambio gaseoso. Es muy probable que el sistema de control tenga mecanismos redundantes que detecten los mismos estímulos, por lo que la ausencia de uno (por ej., cuerpos carótideos) aún da una respuesta normal de steady-state, pero causa variaciones sutiles de la respuesta normal (por ej., cambio en la cinética ventilatoria). Por lo tanto, puede ser necesario apelar a varios mecanismos para determinar la contribución de cada uno en la respuesta integrada que constituye la hiperpnea del ejercicio
CONCLUSION
- Los mecanismos biológicos de control minimizan.
Agradecimientos: Este capítulo fue apoyado, en parte, por una beca del Servicio de Salud Pública (Beca HL-11907).
Referencias
1. Adams, L, B A Cross, H Frankel, R Furneaux, J Garlick, A Guz, K Murphy, and S J G Semple (1980). Effect of Spinal cord transection on steady- state ventilatory response to exercise in man (Abstract). J Physiol. London 308: 63P
2. Adams, L, B A Cross, H Frankel, R Fruneaux, J Garlick, A Guz, K Murphy, and S J G Semple (1981). The dynamics of the ventilatory response to voluntary and electrically induced exercise in man: the influence of the spinal cord (Abstract). J Physiol London 310: 67P
3. Armstrong, B W, H H Hurt, R W Blide, and J M Workman (1961). The humoral regulation of breathing. Science 133: 1897-1906
4. Asmussen, E (1965). Muscular exercise. In: Handbook of Physiology Respiration, edited by W O. Fen and H. Rahn. Washington, DC: Am. Physiol Soc, sect. 3, vol 11, chapter 36, p 939-978
5. Asmussen, E (1973). Ventilation at transition from rest to exercise. Acta Physiol Scand 89: 68-78
6. Asmussen, E, and H Chiodi (1941). The effect of hypoxemia on ventilation and circulation in man. Am J Physiol 132: 426-438
7. Asmussen, E, S H Johansen, M Jorgensen, and M Nielsen (1965). On the nervous factors controlling respiration and circulation duringexercise. Experiments with curarization. Acta Physiol Scand 63: 343-350
8. Asmussen E, and M Nielsen (1958). Pulmonary ventilation and effect of oxygen breathing in heavy exercise. Acta Physiol. Scand. 43: 365378
9. Astrom T. and 0. Wigerts (1966). A Digital Computer for Automatic Breathby-Breath calculation of Respiratory Functions. Stockolm: Karolinska Inst., March, 23p. (Rep. Lab. Aviat. Naval Med.)
10. Aviado D M, Jr., T H Li, W Kalow, C F Schmidt, G L Turnbull, G W Peskin, ME Hess, and A J Weiss (1951). Respiratory and circulatory reflexes from the perfused heart and pulmonary circulation of the dog. Am. J. Physiol. 165: 261-277
11. Bakker H K, R S Struikenkamp, and G A De Vries (1980). Dynamics of ventilation, heart rate, and gas exchange: sinusoidal and impulse work loads in man. J. Appl. Physiol.: Respirat. Environ. Exercise Physiol.48:289-301
12. Band D M, I R Cameron, and S J G Semple (1969). Oscillations in arterial pH with breathing in the cat. J. Appl. Physiol. 26: 261-267
13. Banister E W, and J Griffiths (1972). Blood levels of adrenergic amines during exercise. J. Appl. Physiol. 33: 674-676
14. Bannister R G, and D J C Cunningham (1954). The effects on the respiration and performance during exercise of added oxygen to the inspired air. J. Physiol. London 125: 118-137
15. Banzett R B, H M Coleridge, and J C G Coleridge (1978). Pulmonary CO. ventilatory reflex in dogs: effective range of CO, and results of vagal cooling. Respir. Physiol. 34: 121-134
16. Bartlett D, and G Sant'Ambrogio (1976). Effects of local and systemic hypercapnia on the discharge of strech receptors in the airways of the dog. Respir. Physiol. 26: 91-99
17. Bartoli A, B A Cross, A Guz, S K Jain, M I M Noble, and D Trenchard (1974). The effect of carbon dioxide in the airways and alveoli on ventilation: a vagal reflex studied in the dog. J. Physiol. London 240: 91-109
18. Beaver W L, N Lamarra, and K Wasserman (1981). Breath-by-breath measurement of true alveolar gas exchange. J. Appl. Physiol.: Respirat. Environ. Exercise Physiol. 51: 1662-1675
19. Beaver W L, K Wasserman, and B J Whipp (1973). On-line computer analysis and breath-by-breath graphical display of exercise function tests. J. Appl. Physiol. 34: 128-132
20. Bennett F M, P Reischl, F S Grodins, S M Yamashiro, and W E Fordyce (1981). Dynamics of ventilatory response to exercise in humans. J. Appl. Physiol.: Respirat. Environ. Exercise Physiol. 51: 194-203
21. Bernard T E (1977). Aspects of on-line digital integration of pulmonary gas transfer. J. Appl. Physiol.: Respirat. Environ. Exercise Physiol. 43: 375-378
22. Bessou P, P Dejours, and Y Laporte (1959). Action ventilatoire reflexe de fibres afferentes de grand diametre d origine musculaire chez le chat. J. Physiol. Paris 51: 400-401
23. Bessou P, P Dejours, and Y Laporte (1959). Effects ventilatoires reflexes de la stimulation des fibres afferentes de grand diametre d origine musculaire chez le chat. C. R. Soc. Biol. 153: 477-481
24. Biscoe T J (1977). The carotid body. What next?. Am. Rev. Respir. Dis. 115: 189-191
25. Bisgard G E, H V Forster, B Byrnes, K Stanek, J Klein, and M Manohar (1978). Cerebrospinal fluid acid-base balance during muscular exercise. J. Appl. Physiol.: Respirat. Environ. Exercise Physiol. 45: 94-101
26. Black A M S, N W Goodman, B S Nail, P S Rao, and R W Torrance (1973). The significance of the timing of chemoreceptor impulses for their effect upon respiration. Acta Neurobiol. Exp. Warsz. 33: 139-147
27. Black A M S, and R W Torrance (1971). Respiratory oscillations in chemoreceptor discharge in the control of breathing. Respir. Physiol. 13: 221-237
28. Broman S, and O Wigertz (1971). Transient dynamics of ventilation and heart rate with step changes in work load from different load levels. Acta Physiol. Scand. 81: 54-74
29. Brown H v, K Wasserman, and B J Whipp (1976). Effect of beta-adrenergic blockade during exercise on ventilation and gas exchang. J. Appl. Physiol. 41: 886-892
30. Cardus D, and L Newton (1970). Development of a computer technique for the on-line processing of respiratory variables. Comput. Biol. Med. 1:125-133
31. Casaburi R, R E Dutton, and T Gabriele (1971). An off-line device for breath-by -breath analysis of respiration (Abstract). J. Assoc. Adv. Med. Instrm.5: 107
32. Casaburi R, R W Stremel, B J Whipp, W L Beaver, and K Wasserman (1980). Alteration byhyperoxiaofventilatorydynamics during sinusoidal work. J. Appl. Physiol. Respirat. Environ. Exercise Physiol.48, 1083-1091
33. Casaburi R, M L Weissman, D J Huntsman, B J Whipp, and K Wasserman (1979). Determinants of gas exchange kinetics during exercise in the dog. J. Appl. Physiol.: Respirat. Environ. Exercise Physiol. 46: 1054-1060
34. Casaburi R, B J Whipp, K Wasserman, W L Beaver, and S N Koyal (1977). Ventilatory and gas exchange dynamics in response to sinusoidal work. J. Ap- pl. Physiol. Respirat. Environ. Exercise Physiol. 42: 300-311
35. Casaburi R, B J Whipp, K Wasserman, and S N Koyal (1978). Ventilatory and gas exchange responses to cycling with sinusoidally varying pedal rate. J. Appl. Physiol. Respirat. Environ. Exercise Physiol. 44: 97-103
36. Casaburi R, B J Whipp, K Wasserman, and R W Stremel (1978). Ventilatory control characteristics of the exercise hyperpnea as discerned from dynamic forcing techniques. Chest. 73, Suppl.: 2805-2835
37. Cerretelli P, R Sikand, and L E Farhi (1966). Readjustments in cardiac output and gas exchange during onset of exercise and recovery. J. Appl. Physiol. 21: 1345-1350
38. Cherniack N S, and G S Longobardo (1970). Oxygen and carbon dioxide gas stores of the body. hysiol. Rev. 50: 196-243
39. Clode M, and E J M Campbell (1969). The relationship between gas exchange in blood lactate concentrations during exercise. Ch. Sci. 37: 263-272
40. Coleridge H M, and J C G Coleridge (1964). Cardiac receptors in the dog, with particular reference to two types of afferent endings in the ventricular wall. J. Psysiol London 174:323-339
41. Coleridge H M, J C G Coleridge, and R B Banzett (1978). Effect of CO, on afferent vagal endings in the canine lung. Respir. Physiol. 34: 135-151
42. Coleridge H M, J C G Coleridge, and A Howe (1967). Search forpulmonary arterial chemoreceptors in the cat, with a comparison to the blood supply of the aortic bodies in the newborn and adult animal. J. Physiol. London 191:353-374
43. Comroe J H, Jr (1939). The location and function of the chemoreceptors of the aorta. Am. J. Physiol. 127: 176-191
44. Comroe J H, Jr, and C F Schmidt (1943). Reflexes from the limbs as a factor in the hyperpnea of muscular exercise. Am. J. Physiol. 138:536.547
45. Craig FN, E G Cummings, and W V Blevins (1963). Regulations of breathing at beginning of exercise. J. Appl. Physiol. 18:1183-1187
46. Crandall E D, and J E O'Bransky (1978). Direct evidence of participation of rat lung carbonic anhydrase in CO, reactions. J. Ch. Invest. 62:618-622
47. Cropp G J A, and J H Comroe, Jr (1961). Role of mixed venous blood PCO, in respiratory control. J. Appl. Physiol. 16:1029-1033
48. Cross B A, A Davey, A Guz, P G Katona, M McLean, K Murphy, S J G Semple, and R Stidwell (1982). The role of spinal cord transmission in the ventilatory response to electrically induced exercise in the anesthetized dog. J. Physiol. London 329:37-55
49. Cross B A, A Davey, A Guz, P G Katona, M McLean, K Murphy, S G Semple,and R Stidwell (1982). The pH oscillations in arterial blood during exercise; a potential signal for the ventilatory response in the dog. J. Physiol. London 329:57-73
50. Cross B A, P W Jones K D Leaver, S J G Semple, and R Stidwell (1981). The relationship between CO, output, pulmonary ventilation and the rate of change of arterial pH of the downstroke of the oscillation in the cat (Abstract). J. Physiol. London 320:IOOP-101p
51. Cunningham D J C (1963). Some quantitative aspects of the regulation of human respiration in exercise. Br. Med. Bull. 19:19-25
52. Cunningham D J C (1974). The control system regulating breathing in man. Q. Rev. Biophys. 6:433-483
53. Cunningham DJC (1974). Integrative aspects oftheregulation ofbreathing: a personal view. In: Respiratory Physiology I, edited by J G Widdecombe. Baltimore, Md: University Park, vol. 2, p. 303369. (Int. Rev. Physiol. Ser.)
54. Cunningham D J C (1975). A model illustrating the importance of timing in the regulation of breathing. Nature London 253:440-442
55. Daly W J,and T Overley (1966). Modification of ventilatory regulation by hypnosis. J. Lab. Ch. Med. 68:279-285
56. D'Angelo E, and G Torelli (1971). Neural stimuli increasing respiration during different types of exercise. J. Appl. Physiol. 30:116-121
57. Davis J A, B J Whipp, and K Wasserman (1982). Effect of increment duration on the region of isocapnic buffering during cycle ergometer exercise (Abstract). Physiol. 25(4):252
58. Dawes G S, and J H Comroe, Jr (1954). Chemoreflexs from the heart and lungs. Physiol. Rev. 34:167-201
59. Defares J G (1964). Principles of feedback control and their application to the respiratory control system. In: Handbook of Physiology. Respiration, edited by W O Fenn and H Rahn. Washington, DC:Am. Physiol. Soc, sect.3 vol. 1, chapter 26, P. 649-680
60. Dejours P (1959). La regulation de la ventilation au tours de 1 exercise musculaire chez 1 homme. J. Physiol. Paris 51:163-261
61. Dejours P (1962). Chemoreflexes in breathing. Physiol. Rev. 42:335-358
62. Dejours P (1963). Control of respiration by arterial chemoreceptors. Ann. NY Acad. Sci. 109: 682-695
63. Dejours P (1963). he regulation of breathing during muscular exercise in man: a neuro-humoral theory. In: The Regulation of Human Respiration, edited by D J C Cunningham and B B Loyd. Oxford, UK: Blackwell, p 535-547
64. Dejours P (1964). Control of respiration in muscular exercise. In: Handbook of Physiology. Respiration, edited by W O Fenn and H Rahn. Washington, DC: Am. Physiol. Soc., sect.3, vo1.I, chapt.25, p.631-648
65. Dejours P (1967). Neurogenic factors in the control of ventilation during exercise. Circ. Res. 20/21, Suppl. 1:1146-1153
66. Dejours P, Y Bechtel-Labrousse, and J Raynaud (1961). Etude du controle de la frequence cardiaque et de la ventilation au tours des exercises passifet actif chez l homme. C R Acad Sci Paris 252:2012-2014
67. Dejours P, R Lefrancois, R Flandrois and A Teillac (1960). Autonomic des stimulus ventilatoires oxygene, gaz carbonique et neurogenique de Pexercise musculaire. J Physiol Paris 52:63-64
68. Diamond L B, R Casaburi, K Wasserman, and B J Whipp (1977). Kinetics of gas exchange and ventilation in transitions from rest or prior exercise. J Appl Physiol.: Respirat. Environ. Exercise Physiol. 43:704-708
69. Dolan M, B J Whipp, W Davison, R Weitzman, and K Wasserman (1981). Hypopnea associated with acetate hemodialysis: CO,-flow dependent ventilation in man. N.Engl. J. Med. 305:72-75
70. Douglas C G (1914). Die regulation der atmung beim menschen. Ergeb. Physiol. Biol. Chem. Exp. Pharmakol. 14:338-430
71. Douglas C G (1927). Coordination of the respiration and circulation with variations in bodily activity. Lancet 2:213-218
72. Douglas C G, and J S Haldane (1909). The regulation of normal breathing. J Physiol. London 38:420-440
73. Dutton R E, W A Hodson, D G Davies, and V Chernick (1967). Ventilatory adaptation to a step change in PC02 at the carotid bodies. J. Appl. Physiol. 23:195-202
74. Edwards R H T, D M Denison, G Jones, C T M Davies, and E J M Campbell (1972). Changes in mixed venous gas tensions at start of exercise in man. J. Appl. Physiol. 32:165-169
75. Effros R M (1978). Pulmonary carbonic anhydrase and the release of carbon dioxide from plasma bicarbonate. In: The regulation of respiration during sleep and anesthesia, edited by R S Fitzgerald, H Gautier, and S Lahiri. New York: Plenum, p.255-268
76. Effros R M, R S Y Chang, and P Silverman (1978). Carbonic anhydrase activity of the pulmonary vasculature. Science 199:427-429
77. Eisele J H, B C Ritchie, and J W Severinghaus (1967). Effect of stellate ganglion blockade on the hyperpnea of exercise. J. Appl. Physiol. 22:966-969
78. Eldridge F L (1972). The importance of timing on the respiratory effects of intermittent carotid body chemoreceptor stimulation. J. Physiol. London 222:319-333
79. Eldridge F L (1976). Central neural stimulation of respiration in unanesthetized decerebrate cats. J. Appl. Physiol. 40:23-28
80. Eldridge F L (1976). Expiratory effects of brief carotid sinus nerve and carotid body stimulations. espir. Physiol. 26:395-410
81. Eldridge F L (1977). Maintenance of respiration by central neural feedback mechanisms. Federation Proc. 36:2400-2404
82. Eldridge F L, and P Gill-Kumar (1980). Mechanisms of hyperpnea induced by isoproterenol. Respir. Physiol. 40:349-363
83. Eldridge F L, D E Millhom, and T G Waldrop (1981). Exercise hyperpnea and locomotion: parallel activation from the hypothalamus. Science 211:844-846
84. Engeman R M, G D Swanson, and H Jones (1979). Input desing for model discrimination: application to respiratory control during exercise. IEEE Trans. Biomed.Eng. 26:579-585
85. Filley G F, F Gregoire, and G W Wright (1954). Alveolar and arterial oxygen and the significance of the alveolar-arterial oxygen tension difference in normal man. J. Clin. Invest. 33:517-527
86. Filley G F, and F G Heineken (1976). A blood gas disequilibrium theory. Br. J. Dis. Chest 70:223-245
87. Flandrois R, R Faiven, and J M Pequignot (1977). Role of adrenaline in gas exchanges and respiratory control in the dog at rest and exercise. Respir. Physiol. 30:291-303
88. Flandrois R, J R Lacour, I Islas-Maroquin, and J Chariot (1967). Limb mechanoreceptors inducing the reflex hyperpnea of exercise. Respir Physiol. 2:335-343
89. Forster R E, and E D Crandall (1975). Time course of exchanges between red and extracellularfluid duringCO, uptake. J. Appl. Physiol. 38:710-718
90. Fujihara Y, J R Hildebrandt, and J Hildebrandt (1973). Cardiorespiratory transients in exercising man. 1. Tests of superposition. J. Appl. Physiol. 35: 58-67
91. Fujihara Y, J Hildebrandt, and J R Hildebrandt (1973). Cardiorespiratory transients in exercise man. 11. Linearmodels. J. Appl. Physiol. 35:6876
92. Gardner E, and J Jacobs (1948). Joint reflexes and regulation of respiration during exercise. Am. J. Physiol. 153:567-579
93. Gautier H, A Lacaisse, and P Dejours (1969). Ventilatory response to muscle spindlestimulation by succinylcholine in cats. Respir. Physiol. 7:383-388
94. Geppert J, and N Zuntz (1888). Ueber die Regulation der Atmung. Pluegers Arch. Gesamte Physiol. Menschen Tiere 42:189-245
95. Gonzalez F, Jr, W.E Fordyce, and F S Grodins (1977). Mechanism of respiratory responses to intravenous NaHCO, HCI, and KCN. J Appl. Physiol.: Respirat. Environ. Exercise Physiol. 43:1075-1079
96. Goodwin G M, D I McCloskey, and J H Mitchell (1972). Cardiovascular and respiratory responses to changes in central command during isometric exercise at constant muscle tension. J. Physiol. London 226:173-190
97. Grant B, and S J G Semple (1976). Mechanisms whereby oscilations in arterial carbon dioxide tension might affect pulmonary ventilation. In: Morphology and Mechanisms of chemoreceptors, edited by A S Paintal. Delhi: Univ. Of Delhi Press, p191-196
98. Gray J S (1950). Pulmonary Ventilation and its Physiologucal Regulation. Springfield, IL: Thomas, 82p
99. Greco E C Jr, W E Fordyce, F Gonzalez Jr, P Reischl, and F S Grodins (1978). Respiratory responses to intravenous and intrapulmonary CO, in awake dogs. J. Appl. Ptnysiol.: Respirat. Environ. Exercise Physiol. 45:109-114
100. Griffiths T L, L C Henson, D J Huntsman, K Wasserman, and B J Whipp (1980). The influence of inspired 0, partial pressure on ventilatory and gas echange kinetics during exercise. (Abstract). J. Physiol. London 306:34P
101. Grodins FS (1950). Analysis of factors concerned in regulation of breathing in exercise. Physiol. Rev. 30:220-239
102. Grodins FS (1963). Control Theory and Biological Systems. NY: Columbia Univ. Press, 205p
103. Grodins F S, and G James (1963). Mathematical models of respiratory regulation. Ann. NY Acad. Sci. 109:852-868
104. Guenard H, B Champbell, and D Baergeton (1976). Characteristics of the regulation of respiration during moderate muscular exercise. Acid Base Homeostasis of the Brain Extracellular Fluid and the Respiratory Control System; ed. by Loeschcke. Stuttgart, West Germany: Thime, p.203-209
105. Haggendal J, L H Hartley, and B Saltin (1970). Arterial noradrenaline concentration during exercise in relation to the relative work levels. Scand.J. Clin. Lab. Invest. 26:337-342
106. Haldane J S, and J G Priestley (1905). The regulation of the lung-ventilation. J. Physiol. London 32:225-266
107. Haldane J S, and J G Priestley (1935). Respiration. Oxford, UK: Clarendon, 493p
108. Hall M (1855). Apersu du systeme spinal, ou de la serie des actions reflexes dans leurs applications a la pathologie et specialment a l epilepsie. Paris: Masson, 85p
109. Hansen J E, G P Stelter, and J A Vogel (1967). Arterial pyruvate, lactate, pH, and PCO, during work at sea level and high altitude. J Appl. Physiol. 23: 523-530
110. Harrison T R, W G Harrison Jr, J A Calhoun, and J P Marsh (1932). Congestive heart failure. XVII. The mechanism of dyspnea on exertion. Arch. Intern. Med. 50:690-720
111. Hasselbalch K A, and C Lundsgaard (1912). Blutreaktion and Lungenventilation. Skand. Arch. Physiol. 27:13-32
112. Henderson Y (1938). Adventures in respiration. Baltimore, MD: Williams & Wilkins, 316p
113. HerxheimerH, and R Kost (1928). Das Verhaltnis von Sauerstoffaufnahme and Kohlensaureauusscheidung zur Ventilation bei harter Muskelarbeit. Z. Klin. Med. 108:240-247
114. Heymans C, J J Bouckaert, U S von Euler, and L Dautrebande (1932). Sinus carotidiens et reflexes vaso-moteurs. Arch. Int. Pharmacodyn. Ther. 43:86-110
115. Heymans J F, and C Heymans (1927). Sur les modifications directes et sur la regulation reflexe de l activite du centre respiratoire de la tete isolee du chien. Arch. Int. Pharmacodyn. Ther. 33:273-372
116. Hill E P, G G Power, and R D Gilbert (1977). Rate of pH changes in blood plasma in vitro and in vivo. J. Appl. Physiol.: Respirat. Environ. Exercise Physiol. 42:928-934
117. Hodgson H J F, and P B C Mathews (1968). The ineffectiveness of excitation of the primary endings of the muscle spindle by vibration as a respiratory stimulant in the decerebrate cat. J. Physiol. London 194:555-563
118. Hollmann W, and K Kastner (1969). The behaviour of arterial blood gases, arterial substrates, pH, and haematocrit in different ergometric work. In: Biochemistry of Exercise: Medicine and Sport, edited by J R Poortmans. NY: Karger, vol.3, p.81-88
119. Holmgreen A, and M B Mcllroy (1964). Effect of temperature on arterial blood gas tensions and pH during exercise. J Appl. Physiol. 19: 243-245
120. Honda Y, S Watanabe, I Hashizume, Y Satomura, N Hata, Y Sakakibara, and J W Severinghaus (1979). Hypoxic chemosensitivity in asthmatic patients two decades after carotid body resection. J. Appl. Physiol.: Respirat. Environ. Exercise Physiol. 46:632-638
121. Hornbein T F, S C Sorensen, and C R Parks (1969). Role of muscle spindles in lower extremities in breathing during bicyle exercise. J Appl. Physiol. 27: 476-479
122. Huszczuk A, J Kulesza, and M Ryba (1976). Dependence of total strech receptor activity on airway CO. Bull. Eur. Physiol. Respir. 12:228-229
123. Huszczuk A, A Oren, L E Nery, E Shors, B J Whipp, and K Wasserman (1981). Evidence that venous return, per se, contributes to the exercise hyperpnea (Abstract). Physiologit 24(4):100
124. Iaria C T, U H Jalar, and F F Kao (1959). The peripheral neural mechanism of exercise hyperpnea (Abstract). J. Physiol. London 149:49p-50p
125. Jensen J I (1972). Neural ventilatory drive during arm and leg exercise. Scand. J. Clin. Lab. Invest. 29: 177-184
126. Jensen J I, H Vejby-Christensen, and E S Petersen (1971). Ventilation in man at onset of work employing different standardized starting orders. Respir. Physiol. 13:209-220
127. Jones N L (1975). Exercise testing in pulmonary evaluation: rationale, methods, and the normal respiratory response to exercise. N. Engl. J. Med. 293:541-544
128. Jones P W, W French, M L Weissman, and K Wasserman (1981). Ventilatory responses to cardiac output changes in patients with pacemakers. J.Appl. Physiol.: Respirat. Environ. Exercise Physiol. 51:1103-1107
129. Jones P W, A Huszczuk, and K Wasserman (1982). Cardiac output as a controller of ventilation through changes in right ventricular load. J. Appl. Physiol.: Respirat. Environ. Exercise Physiol. 53:218-224
130. Juratsch C E, A Huszzzuk, aS Gianotta, and B J Whipp (1981). Evidence for a «cardiodynamic» component of the isoproterenol-induced hyperpnea in the dog (Abstract). Federation Proc. 40:567
131. Kaijser L (1970). Limiting factors for aerobic muscle performance. The influence of varying oxygen pressure and temperature. ActaPhysiol. Scnad. Suppl. 436:1-96
132. Kalia M, J M Senapati, B Parida, and A Panda (1972). Reflex increase in ventilation by muscle receptors with nonmedullated fibers (C fibers). J. Appl. Physiol. 32:189-193
133. Kao F F (1963). An experimental study of the pathways involved in exercise hyperpnea employing cross-circulation techniques. In: The Regulation of Human Respiration, edited by D J C Cunningham and B B Lloyd. Oxford, UK: Blackwell, p.461-502
134. Kao F F, C W Wang, S S Mei, and C C Michel (1965). Relationship of exercise hyperpnea to CSF pH: Cerebrospinal Fluid and the Regulation of Ventilation, edited by C McC. Brooks, F F Kao, and B B Lloyd. Oxford, UK: Blackwell, p. 269-275
135. Karlsson H, B Lindborg, and Linnarsson (1975). Time courses of pulmonary gas exchange and heart rate changes in supine exercise. Acta Physiol. Scand. 95: 329-340
136. Karlsson H, and O Wigertz (1971). Ventilation and heart-rate responses to ramp-fuction changes in work load. Acta Physiol. Scand. 81:215-224
137. Kellogg R H (1981). Historical perspectives. In: Regulation of Breathing, edited by Hornbein. NY: Dekker, p21
138. Keul J, E Doll, and D Keppler (1972). Energy Metabolism of Human Muscle. Baltimore, MD: University Park, p. 71-78
139. Klocke R A (1978). Catalysis of C02 reactions by lung carbonic anhydrase. J. Appl. Physiol.: Respirat. Environ. Exercise Physiol. 44:882-888
140. Kiozumi K, J Ushiyama, and McM. Brooks (1961). Muscle afferents and activity of respiratory neurons. Am J Physiol. 200:679-684
141. Kostreva D R, F A Hopp, E J Zuperku, and J P Kampine (1979). Apnea, tachypnea, and hypotension elicited by cardiac vagal afferents. J Appl. Physiol. Respirat. Environ. Excercise Physiol. 47:312-318
142. Kostreva D R, E J Zuperku, R V Purtock, R L Coon, and J P Kampine (1975). Sympathetic afferent nerve activity of right heart origin. Am. J Physiol. 229: 911-915
143. Krogh A, and J Linhard (1913). The regulation of respiration and circulation during the initial stages of muscular work. J. Physiol. London 47:112-113
144. Lahiri S, and R G Delaney (1975). Stimulus interaction in the responses of the carotid body chemoreceptors single afferent fibers. Respirat. Physiol.24: 249-266
145. Lamb T W (1968). Ventilatory response to hind limb exercise in anesthetized cats and dogs. Respir. Physiol. 6:88-104
146. Lamb T W, N R Anthonisen, and S M Tenney (1965). Controlled frequency breathing during muscular exercise. J. Appl. Physiol. 20:244-248
147. Lambertsen C J,and R Gelfand (1966). Breath-by-breath measurement of respiratory functions: instrumentation and applications. J. Appl. Physiol. 21:282-290
148. Ledsome I R (1977). The reflex role of pulmonary arterial baroreceptors. Am. Rev. Resp. Dis. 115:245-250
149. Leitner L M, and P Dejours (1971). Reflex increase in ventilation induced by vibrations of the triceps surae muscles in the cat. Respir. Physiol. 12:199-204
150. Leusen I (1965). Aspects of the acid-base balance between blood and cerebrospinal fluid. In: Cerebrospinal Fluid and the Regulation of Ventilation, edited by C McM.Brooks, F F Kao, and B B Lloyd. Oxford, UK: Blackwell, p. 55-89
151. Levine S (1978). Ventilatory response to muscularexercise. In: Regulation of Ventilation and Gas Exchange, edited by D G Davies and C D Barnes. NY: Academic, p.31-68
152. Lewis S M (1975). Awake baboon s ventilatory response to venous and inhaled CO, loading. J. Appl. Physiol. 39:417-422
153. Liang C S, and W B Hood Jr (1976). Afferent neural pathway in the regulation of cardiopulmonary responses to tissue hypermetabolism. Circ. Res. 38:209-214
154. Linnarsson D (1974). Dynamics of pulmonary gas exchange and heart rate changes at start and end of exercise. Acta Physiol. Scand. Suppl. 415:1-68
155. Linnarson D, and B Lindborg (1974). Breath-by-breath measurement of respiratory gas exchange using on-line analog computation. Scand. J. Clin. Lab. Invest. 34:219-224
156. Linton R A F, R Miller, and I R Cameron (1976). Ventilatory response to C02 inhalation and intravenous infusion of hypercapnic blood. Respir. Physiol. 26: 383-394
157. Liu C T, R A Huggins, and H E Hoff (1969). Mechanisms of intra-arterial K induced cardiovascular and respiratory responses. Am. J. Physiol. 217:969-973
158. Lloyd B B, and J M Patrick (1963). Respiration and metabolic rate during active and pasive exercise (Abstrac). J. Physiol. London 165:67p 68p
159. Loeppky J A, E R Greene, D E Hoekenga, A Caprihan, and U C Luft (1981). Beat-by-beat stroke volume assessment by pulsed Doppler in upright and supine exercise. J. Appl. Physiol.: Respirat. Environ.Exercise Physiol. 50:1173-1182
160. Lugliani R, B J Whipp, C Seard, and K Wasserman (1971). Effect of bilateral carotid-body resection on ventilatory control at rest and during exercise in man. N. Engl. J. Med. 285:1105-1111
161. Marsh J and P G C Nye (1981). A simple method of venous carbon dioxide loading (Abstract). J. Physiol. London 312:56p
162. Marsh R H K, K R Lyen, G A D McPherson, S B Pearson, and D J C Cunningham (1973). Breath-by-breath effects of imposed alternatebreath oscillations of alveolar CO2. Respir. Physiol. 18:80-91
163. Masson R G, and S Lahiri (1974). Chemical control of ventilation during hypoxic exercise. Respir. Physiol. 22:241-262
164. Matell G (1973). Time-course of changes in ventilation and arterial gas tensions in man induced by moderate exercise. Acta. Physiol. Scand. Sppl. 206:1-53
165. McCloskey D I, and J H Mitchel (1972). Reflex cardiovascularand respiratory responses originating in exercising muscle. J. Physiol. London 224:173-186
166. Milhorn H T (1966). The Application of Control Theory to Physiological Systems. Philadelphia, PA:Saunders, 386 p
167. Miyamoto Y, T Hiura, T Tamura, T Nakamura, J Higuchi, and T Mikami (1982). Dynamics of cardiac, respiratory, and metabolic function in men in response to step work load. J. Appl. Physiol.: Respirat. Environ. Exercise Physiol. 52:1198-1208
168. Moore R M, R E Moore, and A O Singleton Jr (1934). Experiments on the chemical stimulation of pain-endings associated with small blood vessels. Am. J. Physiol. 107:594-602
169. Morgan W P, P B Raven, B L Drinkwater, and S M Horvath (1973). Perceptual and metabolic responsivity to standard bicycle ergometry following various hypnotic suggestions. Int. J. Clin. Exp. Hypn. 21:86-101
170. Mustafa M E, and M J PurveS (1972). The effect of CO, upon discharge from slowly adapting strech receptors in the lungs of rabbits. Respir. Physiol. 16: 197-212
171. Naimark A, K W asserman,and M B Mcllroy (1964). Continous measurement of ventilatory exchange ratio during exercise. J. Appl. Physiol. 19:644-652
172. Nery L E, K Wasserman, J D Andrews, D J Huntsman, J E Hansen, and B J Whipp (1982). Ventilatory and gas exchange kinetics during exercise in chronic airways obstructioN. J. Appl. Physiol.: Respirat. Environ. Exercise Physiol. 53:1594-1062
173. Nims L F, and C Marshall (1938). Blood pH in vivo. I. Changes due to respiration. Yale J. Biol. Med. 10:445-448
174. Nye P C G, J Marsh, and R A Briggs (1981). Venous C02 loading via the gut as a stimulus to ventilation and cardiac output (Abstract). Federation Proc. 40:482
175. Ochwadt B, E Bucherl, H Kreutzer, and H H Loeschcke (1959). BeeinfluussungderAtemsteigerungbeiMuskelarbeitdurchpartiellen neuro-muskularen Block (Tubocurarin). Pflegers Arch. Gesamme Physiol. Menchen Tiere 269:613-621
176. Oren A, K Wasserman, J A Davis, and B J Whipp (1981). Effect of CO, set point on ventilatory response to exercise. J. Appl. Physiol.: Respirat Environ. Exercise Physiol. 51:185-189
177. Oren A, B J Whipp, and K Wasserman (1982). Effect of acid-base status on the kinetics of the ventilatory response to moderate exercise. J Appl. Physiol.: Respirat. Environ. Exercise Physio152:1013-1017
178. Paulev, P E (1971). Respiratory and cardiac responses to exercise in man. J Appl. Physiol. 30: 165-172
179. Pearce, D H, H T Milhorn, Jr (1977). Dynamic and steady-state respiratory responses to bicycle exercise. J Appl. Physiol.: Respirat. Environ. Exercise Physiol. 42: 959-967
180. Pearce, D H, H T Milhorn, Jr, G H Holloman, Jr, and W J Reynolds (1977). Computer-based system for analysis of respiratory responses to exercise. J Appl. Physiol.: Respirat. Environ. Exercise Physiol. 42: 968-975
181. Petersen, E S, B J Whipp, J A Davis, D J Huntsman, H V Brown, and K Wasserman (1983). Effects of a adrenegic blockade on ventilation and gas exchange during exercise in humans. J Appl. Physiol.: Respirat. Environ. Exercise Physiol. 54: 1306-1313
182. Petersen, E S, B 1 Whipp, D B Drysdale, and D J C Cunningham (1978). The relation between arterial blood gas oscillation in the carotid region and the phase of the respiratory cycle during exercise in man: testing a model. In: The Regulation of Respiration During Sleep and Anesthesia, edited by R S Fitz-gerald, H Gautier, and S Lahiri. New York: Plenum, p. 335-342
183. Phillipson, E A, J Duffin, and J D Cooper (1981). Critical dependence of respiratory rhythmicity on metabolic CO, load. J Appl. Physiol: Respirat. Environ Exercise Physiol. 50: 45-50
184. Ponte, J and M J Purves (1978). Carbon dioxide and venous return and their interaction as stimuli to ventilation in the cat. J Physiol London 274: 455-475
185. Purves, M J (1966). Fluctuations of arterial oxygen tension which have the same period as respiration. Respir. Physiol. 1: 281-296
186. Rahn, H, and W O Fenn (1955). A graphical analysis of the respiratory gas exchange. The 0= CO2 Diagram. Washington, DC: Am. Physiol. Soc., 43 p
187. Ramsay, A G (1955). Studies on the Control of Respiration. Glasgow, UK: Univ. of Glasgow Press, p. 84-114. PhD thesis
188. Ramsay, A G (1959). Effects of metabolism and anesthesia on pulmonary ventilation. J Appl Physiol 14: 102-104
189. Rasmussen, B, K Klausen, J P Clausen, and J Trap-Jensen (1990). Pulmonary ventilation, blood gases, and blood pH after training of the arms and legs. J Appl. Physiol. 38: 250-256
190. Riley, R L (1963). The hyperpnea of exercise. In: The Regulation of Human Respiration, edited by D J C Cunningham and B B Lloyd. Oxford, UK: Blackwell, p 525-534
191. Saltzman, H A, and H O Sicker (1968). Intestinal response to changing gaseous environments: normobaric and hyperbaric observation. Ann NY Acad Sci. 150: 31-39
192. Saunders, K B (1980). Oscillations of arterial CO, tension in a respiratory model: some implications for the control of breathing in exercise. J Theor Biol 84: 163-179
193. Scherrer, M (1969). Acid-base imbalance and gas exchange during heavy work. In: Biochemestryof Exercise: Medicine and Sport, edited by J.R. Poortmans. New York: Karger, vol 3, p. 2-14
194. Schoener, E P, and H M Frankel (1972). Effect of hyperthermia and PaC02 on the slowly adapting pulmonary stretch receptor. Am J Physiol. 222: 68-72
195. Senapati, J M (1966). Effect of stimulation of muscle afferents on ventilation of dogs. J Apll Physiol. 21:242-246
196. Sheldon, M I, and J F Green (1982). Evidence for pulmonary C02 chemosensitivity: effects on ventilation. J Appl Physiol: Respirat. Environ. Exercise Physiol. 52: 1192-1197
197. Shors, E C, A Huszczuk, K Wasserman, and B J Whipp (1980). Ventilatory responses to venous CO, unloading during steady-state exercise in the dog (Abstract). Federation Proc. 39:583
198. Sipple, J H, and R Gilbert (1996). Influence of proprioceptor activity in the ventilatory response to exercise. J Appl Physiol 21: 143-146
199. Stockley, R A (1978). The contribution of the reflex hypoxic drive to the hyperpnea of exercise. Respir. Physiol. 35: 79-87
200. Stremel, R W, D J Huntsman, R Casaburi, B J Whipp, and K Wasserman (1978). Control of ventilation during intravenous CO, loading in the awake dog. J Appl Physiol: Respirat. Environ. Exercise Physiol. 44: 311-316
201. Stremel, R W, B J Whipp, R Casaburi, D J Huntsman, and K Wasserman (1979). Hypopnea consequent to reduced pulmonary blood flow in the dog. J Appl Physiol: Respirat. Environ. Exercise Physiol. 46: 1171-1177
202. Sue, D Y, J E Hansen, M Blais, and K Wasserman (1980). Measurement and analysis of gas exchange during exercise using a programmable calculator. J Appl. Physiol: Respirat. Environ. Exercise Physiol. 49: 456-461
203. Sue, D Y, L R Van Meter, J E Hansen, and K Wasserman (1983). Exercise gas exchange in asthmatics after B-adrenergic blockade. J Appl Physiol: Respirat. Environ. Exercise. Physiol. 55: 529-533
204. Sutton, J R, N L Jones, and C J Toews (1976). Growth hormone secretion in acid-base alterations at rest and during exercise. Clin. Sci. Mol. Med. 50:241-247
205. Swanson G D (1978). The exercise hyperpneadilemma. Chest 73, Suppl.:277-279
206. Swanson G D (1980). Breath-to-breath considerations for gas exchange kinetics. In: Exercise Bioenergetics and Gas Exchange, edited by P. Cerretelli and B J Whipp. Amsterdam: Elsevier, p.211-222
207. Swanson G D, D S Ward, and J W Bellville (1976). Posthyperventilation isocapnic hyperpnea. J. Appl. Physiol. 40:592-596
208. Swanson G D, B J Whipp, R D Kaufman, K A Aqleh, B Winter, and J W Bellville (1978). Effect of hypercapnia on hypoxic ventilatory drive in carotid body-resected man. Appl. Physiol.: Respirat. Environ Exercise Physiol. 45: 971-977
209. Silvester J T, B J Whipp, and K Wasserman (1973). Ventilatory control during brief infusions of C02 laden blood in the awake dog. J Appl. Physiol. 35:178-186
210. Tawadrous F D, and F L Eldridge (1974). Posthyperventilation breathing patterns after active hyperventilation in man. J. Appl. Physiol. 37:353-356
211. Tibes U (1977). Reflex inputs to the cardiovascular and respiratory centers from dynamically working canine muscles. Some evidence for involvement of group III or IV nerve fibers. Circ. Res.41:332-341
212. Tibes U, B Hemmer, and D Boning (1977). Heart rate and ventilation in relation to venous [K], osmolality, pH, PCO PO [orthophosphate], and [lactate] at transition in athletes and non-athletes. Eur. J. Appl. Physiol. 36:127-140
213. Twentyman O P, A Diesley, H R Gribbin, K G M M Alberti, and A E Tattersfield (1981). Effect 19 adrenergic blockade on respiratory and metabolic responses to exercise. J. Appl. Physiol.: Respirat. Environ. Exercise Physiol. 51: 788-793
214. Uchida Y (1975). Afferent sympathetic nerve fibers with mechanoreceptors in the right heart. Am. J. Physiol. 228:223-230
215. Uchida Y (1976). Tachypneaafter stimulation of afferent cardiac sympathetic nerve fibers. Am. J. Physiol. 230:1003-1007
216. Van Benthuysen K M, G D Swanson, and J V Weil (1980). Role of venous CO, flow in exercise hyperpnea (Abstract). Federation Proc. 39:584
217. Volkman A W (1841). Ueberdie Bewegungen des Athmens and Schluckens, mit besonderer Berucksichtigung neurologisher Streitfragen. Arch. Anat. Physiol. 332-336
218. Von Euler U S, and S Hellner (1952). Excretion of noradrenaline and Kdrenaline in muscular work. Acta. Physiol. Scand 26:183-191
219. Wade J G, C P Larson Jr, R F Hickey, W K Ehrenfeld, and J W Severinghaus (1970). Effect of carotid endarterectomy on carotid chemoreceptor and baroreceptor funtion in man. N. Engl. J. Med. 282:823-833
220. Ward S A (1979). The effects of sudden airway hyperpnia on the initiation of exercise hyperpnea in man. J. Physiol. London 296:203-214
221. Ward S A, and D J C Cunningham (1977). The relation between hypoxia and C02 induced reflex alteration of breathing in man. Respir. Physiol. 29:363-378
222. Ward S A, J A Davis, M L Weissman, K Wasserman, and B J Whipp (1979). Lung gas stores and the kinetics of gas exchange during exercise (Abstract). Physiologist 22(4):129
223. Ward S A, B J Whipp, S Koyal, and K Wasserman (1983). Influence of body CO, stores on ventilatory dynamics during exercise. J. Appl. Physiol.: Respirat Environ. Exercise Physiol. 55:742-749
224. Wasserman K (1976). Testing regulation of ventilation with exercise. Chest 70, Sppl.: 173-178
225. Wasserman K (1978). Breathing during exercise. N. Engl. J. Med. 298:780-785
226. Wasserman K, A L Van Kessel, and G G Burton (1967). Interaction of physiological mechanisms during exercise. J. Appl. Physiol. 22:71 85
227. Wasserman K., and B J Whipp (1975). Exercise physiology in health and disease. Am. Rev. Respir. Dis. 112:219-249
228. Wasserman K,and S J Whipp (1976). The carotid bodies and respiratory control in man. In:Morphology and Mechanisms of Chemorreceptors, edited by A S Paintal. Delhi: Iniv. of Delhi Press, p. 156-175
229. Wasserman K, B J Whipp, RR Casaburi, and W L Beaver (1977). C02 flow and exercise hyperpnea: cause and effect?. Am.Rev. Respir. Dis. 115:225-237
230. Wasserman K, B J Whipp, R Casaburi, W L Beaver, and H V Brown (1977). C02 flow to the lungs and ventilatory control. In: Muscular Exercise and the Lung, edited by J A Dempsey and C E Reed. Madison: Univ. of Wisconsin Press, p. 103-135
231. Wasserman K, B J Whipp, R Casaburi, M Golden, and W L Beaver (1979). Ventilatory control duringexerciseinman. Bull. Eur.Physiopathol. Respir. 15:27-51
232. Wasserman K. B J Whipp, R Casaburi, D J Huntsman, J Castagna, and R Lugliani (1975). Regulation of arterial PC02 during intravenous C02 loading. J. Appl. Physiol. 38:651-656
233. Wasserman K, B J Whipp, and J Castagna (1974). Cardiodynamic hyperpnea: hyperpnea secondary to cardiac output increase. J. Appl. Physiol. 36:457-464
234. Wasserman K, B J Whipp, and J A Davis (1981). Respiration in exercise. In: Respiratory Physiology III, edited by J G Widdicombe. p.149-211. (Int. Rev. Physiol. Ser.) Baltimore, MD: Univ. Park, vol. 3
235. Wasserman K, B J Whipp, S N Koyal, and W L Beaver (1973). Anaerboic threshold and respiratory gas exchange during exercise. J. Appl. Physiol. 35:236-243
236. Wasserman K, B J Whipp, S N Koyal, and M G Cleary (1975). Effect of carotid body resection on ventilatory and acid-base control during exercise. J. Appl. Physiol. 39:354-358
237. Wasserman K, B J Whipp, A Oren, and R Casaburi (1981). Humoral control of ventilation during exercise in man. In: Advances in Physiological Sciences. Respiration, edited by 1. Hutas and L A Debreczeni. Budapest: Akad. Kiado, vol.l0,p.285-297
238. Weiler-Ravell D, D M Cooper, B J Whipp, and K Wasserman (1983). Control of breathing at the start of exercise as influenced by posture. J. Appl. Physiol.: Respirat. Environ. Exercise Physiol. 55:1460-1466
239. Weissman M L, K Wasserman, D J Huntsman, and B J Whipp (1979). Ventilation and gas exchange during phasic hindlimb exercise in the dog. J. Appl. Physiol.: Respirat Environ. Exercise Physiol. 46:878-884
240. Weissman M L, B J Whipp, D J Huntsman, and K Wassserman (1980). Role of neural afferents from working limbs in exercise hyperpnea. J. Appl. Physiol.: Respirat. Environ. Exercise Physiol. 49: 239-248
241. Welch H G, J P Mullin, G D Wilson, and J Lewis (1974). Effects of breathing 0, enriched mixtures on metabolic rate during exercise. Med.Sci. Sports 6: 26-32
242. Whipp B J (1977). The hyperpneaofdynamic muscular exercise. Exercise Sport Sci. Re. 5:295-311
243. Whipp J B (1978). Tenets of the exercise hyperpnea and their degree of corroboration. Chest 73, uppl.:274-277
244. Whipp J B (1981). The control of the exercise hyperpnea. In: The Regulation of breathing, edited by T F Hornbein. New York: Dekker, p.1069-1139
245. Whipp B J, J A Davis, F Torres, and K Wasserman (1981). A test to determine parameters of aerobic funtion during exercise. J. Appl. Physiol.: Respirat. Environ. Exercise Physiol. 50:217-221
246. Whipp B J, and M Mahler (1980). Dynamics of gas exchange during exercise. In: Pulmonary Gas Exchange, edited by J B West. New York: Academic, vol. 2, p 33-96
247. Whipp B 1, J T Sylvester, C Seard, and K Wasserman (1971). Intra-breath respiratory responses following the onset of cycle ergometerexercise. In: Lung Function and Work Capacity. (Symposia proceedings.) Salford, UK: Univ. of Salford Press, p 45-64
248. Whipp, B J, and S A Ward (1980). Control of ventilatory dynamics during exercise. Int. J Sports Med. 1: 146-159
249. Whipp, B J, S A Ward, N Lamarra, J A Davis, and K Wasserman (1982). Parameters of ventilatory and gas exchange dynamics during exercise. J AppI Physiol.: Respirat. Environ. Exercise Physiol. 52:1506-1513
250. Whipp, B J, and K Wasserman (1969). Alveolar-arterial gas tension differences during graded exercise. J Appl Physiol. 27: 361-365
251. Whipp, B J, and K Wasserman (1980). Carotid bodies and ventilatory control dynamics in man. Federation Proc. 39: 2668-2673
252. Whipp, B J, and K Wasserman, R Casaburi, C Jurtsch, M L Weissman, and R W Stremel (1978). Ventilatory control characteristics of condition resultingin isocapnichyperpnea. In:TheRegulation ofRespiration During Sleep and Anesthesia, edited by R S Fitzgerald, H Gautier, and S Lahiri. New York: Plenum, p 355-365
253. Whipp, B J, K Wasserman, J A Davis, N Lamarra, and S A Ward (1980). Determinants of O2 and CO, kinetics during exercise in man. In: Exercise Bioenergetics and Gas Exchange, edited by P. Cerretelli and B J Whipp. Amsterdam: Elsevier, p 175-185
254. Wigertz, O (1970). Dynamics of ventilation and heart rate in response to sinusiodal work load in man. J Appl Physiol 29: 208-218
255. Wigertz, O (1971). Dynamics of respiratory and circulatory adaptation to muscular exercise in man. A systems analysis approach. Acta Physiol Scand Suppl 363: 1-32
256. Wilmore, J H, J A Davis, and A C Norton (1976). An automated system for assessing metabolic and respiratory function during exercise. J Appl Physio140: 619-624
257. Wilson, G D, and H G Welch (1975). Effects of hyperoxic gas mixtures on exercise tolerance in man. Med Sci Sports 7: 48-52
258. Winn, R, J R Hildebrandt, and J Hildebrandt (1979). Cardiorespiratory responses following isoproterenol injection in rabbits. J Appl Physiol: Respirat Environ. Exercise Physiol 47: 352-359
259. Winterstein, H (1911). Die Regulierung de Atmung durch das Blut. Arch. Gesamte Physiol. 138: 167-184
260. Yarnamoto, W S (1960). Mathematical analysis of the time course of alveolar CO2. J Appl Physiol 15: 215-219
261. Yamamoto, W S (1962). Transmission of information by the arterial blood stream with particular reference to carbon dioxide. Biophys J 2:143159
262. Yamamoto, W S and M W Edwards, Jr (1960). Homeostasis of carbon dioxide during intravenous infusion of carbon dioxide. J Appl Physiol 15: 807-818
263. Yokota, H, and F Kreuzer (1973). Alveolar to arterial transmission of oxygen fluctuations due to respiration in anesthetized dogs. Pfluegers Arch 340: 291-306
264. Young, I H, and A J Woolcock (1978). Changes in arterial blood gas tensions during unsteady-state exercise. J Appl Physiol: Respirat. Environ. Exercise Physiol 4: 93-96