
Detección de Eventos Adversos en la Investigación de Suplementación Dietaria: Lecciones de los Alcaloides de la Efedra
1Division of Drug Delivery and Disposition, School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-7360.
2Department of Exercise Science and Athletics, Bloomsburg University, Bloomsburg, PA 17815.
Artículo publicado en el journal PubliCE, Volumen 0 del año 2004.
Publicado 7 de julio de 2004
Resumen
Palabras clave: efedrina, ma huang, efectos colaterales, seguridad, pruebas clínicas
INTRODUCCION
Las definiciones y condiciones legales de la “suplementación dietaria” (e.g., productos a base de hierbas, nutrientes simples aislados) varían dependiendo del país en particular. Algunos países tienen normas rigurosas en cuanto a la regulación y control de calidad de este tipo de productos. Por ejemplo, Alemania tiene directivas bien establecidas mediante su Comisión E, una división independiente de la Agencia de Salud de Alemania Federal. En el Reino Unido, la propuesta “Directiva sobre los Productos Medicinales Tradicionales a base de Hierbas” exige a las compañías que demuestren que el producto a base de hierbas patrocinado no es nocivo para la condición específica que intentan tratar (www.mca.gov.uk). Contrariamente, Estados Unidos considera a los suplementos dietarios en una clasificación independiente a la de los alimentos y las drogas y por esta razón los suplementos dietarios no son evaluados por la Administración de Alimentos y Drogas (FDA) excepto por las afirmaciones de contenido. Con la excepción de nuevos ingredientes dietarios, los patrocinadores no necesitan proveerle a la FDA la evidencia que respalda la seguridad o efectividad de los suplementos, antes o después de la comercialización (42). A diferencia de otros países en Estados Unidos, este tipo de productos no está destinado al diagnostico, tratamiento, cura o prevención de enfermedades. Existe una considerable variabilidad global en la regulación de los suplementos dietarios, y el lector es dirigido a otras publicaciones en la materia (7, 18, 53, 72). En Estados Unidos hay un considerable debate político respecto de la regulación de los suplementos dietarios que surgió del Decreto de Educación Sanitaria de Suplementos Dietarios (DSHEA) de 1994. Este debate se extiende desde problemas concernientes con el control de calidad a la seguridad/toxicología, a si los suplementos dietarios deben ser sometidos a regulaciones más estrictas. Los fabricantes de suplementos y sus grupos de abogados pujan por condiciones de regulación menos estrictas que las actualmente establecidas por la DSHEA, pero algunas organizaciones médicas y farmacéuticas, así como también algunos grupos de abogados públicos, demandan una regulación más estricta y grandes modificaciones a la DSHEA.
El mercado mundial de suplementos dietarios ha crecido, en parte, debido a la creencia de que los productos a base de hierbas están libres de efectos colaterales (18). Sin embargo, esta parece ser una suposición sin fundamentos dado que muchas drogas modernas eran/son derivadas a partir de plantas (e.g., digitalina/digoxina, taxanos para quimioterapia). Además, los países con normas reguladoras bien establecidas han retirado del mercado varios productos a base de hierbas debido a la preocupación por la seguridad. (47). Aunque se han establecido agencias reguladoras, la seguridad es todavía un tema en desarrollo (47). En Estados Unidos hay una preocupación por la emisión de informes de seguridad relacionados a la ingesta de varios suplementos dietarios (e.g., efedra y eventos cardiovasculares; Kava y daño hepático; Mosto de San Juan e interacción con distintas drogas) (31, 39, 55). Desafortunadamente se desconoce la frecuencia y severidad de los eventos adversos asociados con la suplementación dietaria (68).
Aunque en comparación con las drogas modernas los suplementos dietarios pueden no poseer una acción farmacológica inmediata o fuerte, estos tienen la capacidad de modular la fisiología, y por ello poseen propiedades farmacológicas. Por ello, parece apropiado discutir la seguridad de los suplementos dietarios en el contexto de la seguridad de las drogas modernas y utilizar las normas establecidas por la industria farmacéutica, cuerpos gubernamentales tales como la FDA de los Estados Unidos y la “Conferencia Internacional de Armonización” (ICH) (la Conferencia Internacional de Armonización es un proyecto que reúne a las autoridades reguladoras de Japón, Europa y Estados Unidos, y a expertos de la industria farmacéutica, para discutir aspectos científicos y técnicos para el registro de productos farmacéuticos para uso humano) para su aplicación en la investigación sobre suplementos dietarios. Revisiones previas se han enfocado en la regulación global de los suplementos dietarios, las posiciones y definiciones actuales, y los pro y contra de cada cuerpo gubernamental (9, 18, 72). El propósito de esta revisión es discutir los principios generales relacionados a los estudios que enfocan el tema de la seguridad de los suplementos dietarios, utilizando como guía a la industria farmacéutica. Hemos utilizado al suplemento efedra (Ma Huang) como un modelo para ilustrar las limitaciones actuales de la investigación y ofrecer sugerencias para el desarrollo de estudios que puedan examinar rigurosamente los temas relacionados con la seguridad con respecto a los suplementos nutricionales.
EFEDRA (Ma Huang)
La Ephedra es una hierba (e.g. efedra sinica) que contiene una mezcla compleja de alcaloides incluidos la efedrina, la pseudo efedrina, norefedrina (fenilpropanolamina) y metil efedrina (35, 37). La efedrina es una droga imita la acción del sistema nervioso y es un estimulante del sistema nervioso central (41), que se utiliza para la pérdida de peso debido a sus propiedades termogénicas y anorexígenas (5, 22). De este modo, los suplementos a base de efedrina, efedra, y efedra/cafeína son comercializados para la perdida de peso y como estimulantes del rendimiento deportivo. Sin embargo, los reportes de eventos adversos han puesto en duda la seguridad de estos productos (38, 61).
La prevalencia exacta del uso de efedra es desconocida, especialmente a nivel mundial. Sin embargo, los datos de una encuesta realizada en los Estados Unidos indican que la utilización de efedra es del 1% en la población general (11), del 3.5% entre atletas universitarios (33), y del 13 al 25% en miembros de centros de actividad física (46). Debe señalarse que el Reino Unido y algunos países de Europa ya han restringido la utilización de efedra en los remedios a base de hierbas (www.mca.gov.uk). Nosotros utilizamos a la efedra como un ejemplo a lo largo de este artículo debido al beneficio de la estandarización de los alcaloides (34, 37), a que se conoce el período de tiempo de disposición de la droga (43), a los reportes de efectos adversos comunes (3, 13, 14), a los reportes de eventos adversos serios, pero más raros (38, 64), y debido a que la efedrina es un una droga aprobada por la FDA de la cual se conoce sus efectos colaterales e interacción con otras drogas.
Definiendo la Seguridad
El tiempo promedio para el desarrollo de una droga (desde su descubrimiento hasta su aprobación) es aproximadamente 10-15 años (aproximadamente la mitad en pruebas clínicas), lo que implica investigar la seguridad y la eficacia en miles de sujetos, con costos asociados que se aproximan a los US$ 800 millones (60). Sin embargo, la mayor proporción de los US$ 800 millones se gasta en la Fase III, estudios de eficacia a gran escala, mientras que en comparación con estos, los estudios iniciales sobre seguridad y tolerancia son mucho menores y más baratos. Los estudios diseñados para investigar la seguridad de los suplementos dietarios a menudo resultan insignificantes en comparación con los estudios iniciales sobre la tolerancia a la droga y no están diseñados adecuadamente para detectar siquiera los más frecuentes asuntos de seguridad. Por ejemplo, un estudio reciente sobre los efectos cardiovasculares de la suplementación con efedra/cafeína no mostró efectos sobre la frecuencia cardiaca y la presión sanguínea (45). Sin embargo, se han observado incrementos en la presión sanguínea y en la frecuencia cardiaca luego de la ingestión de efedra/cafeína tanto con dosis altas (13, 14) como con dosis bajas (39). Estas discrepancias demuestran la necesidad de condiciones más rigurosas en los estudios de investigación sobre suplementación dietaria diseñados para valorar la seguridad, como es típico en la fase temprana de la investigación con drogas.
Utilizando a la industria farmacéutica como guía, las drogas en desarrollo son consideradas seguras hasta que se pruebe lo contrario, a través de evaluaciones pre clínicas (cultivos celulares, estudios con animales) y clínicas (estudios en humanos) patrocinadas por la industria. La responsabilidad de demostrar la seguridad (y la eficacia) esta puesta en la industria patrocinadora y no en el gobierno. Contrariamente, en Estados Unidos, existe muy poca obligación por parte de la industria patrocinadora del suplemento para probar la seguridad del producto con la cantidad de dosis comercializada al público. Esta falta de evaluación sobre la seguridad de la droga transfiere la responsabilidad a las investigaciones patrocinadas por el gobierno y las universidades. En Alemania bajo el Acta de Medicamentos Alemanes, son necesarios datos bibliográficos (i.e., artículos de revisión bien documentados, pruebas clínicas, y estudios experimentales) para valorar la eficacia y la seguridad de los productos (47). Además, la vigilancia farmacológica de sus productos herbarios es un elemento básico del mercado y una evaluación sistemática de los riesgos debe incluir una prueba de eficacia plausible (47). Cuando se discute la seguridad (sea de una droga o de un suplemento) es importante entender que “seguro” no significa no nocivo, pero significa que los beneficios terapéuticos pesan más que los riesgos para la población a la cual está destinada el producto. Como un ejemplo, los agentes quimioterapéuticos (e.g., etopsido, taxol, vincristina, etc.) pueden tener efectos colaterales comunes (e.g., nauseas, vómitos, alopecia, mielosupresión, molestias gastrointestinales) que son inaceptables en tipos de drogas para enfermedades que no presentan riesgo de muerte. La tolerancia para los riesgos (efectos colaterales) es mayor en productos para las condiciones de amenaza para la vida que en enfermedades que no amenazan la vida.
Un suplemento dietario podría ser clínicamente útil si los efectos terapéuticos deseados (e.g., incremento del vigor, incremento de la sensación de bienestar, incremento de la fuerza muscular, disminución de sentimientos de depresión) pesan más que los efectos farmacológicos/toxicológicos no deseados. Por ejemplo, la efedrina/cafeína y la efedra/cafeína herbaria, junto con modificaciones de la conducta en sujetos obesos (dieta, dieta y ejercicio), causa un incremento ligeramente mayor en la perdida de peso (<3.5 kg en 6 meses) en comparación con la modificación de la conducta por si sola (3, 13, 14, 17). Sin embargo, es discutible si los beneficios terapéuticos (una modesta perdida de peso) son mayores que los riesgos de aparición de los efectos colaterales reportados para la efedra y las mezclas de efedra, tanto de los comunes (incremento de la presión sanguínea y de la frecuencia cardiaca) como de los no tan comunes (infarto de miocardio, accidente cerebro-vascular).
La relación riesgo/beneficio en drogas aprobadas para la perdida de peso también existe, y ha limitado la utilidad terapéutica de medicamentos para la perdida de peso que actúan de manera similar. En el pasado, la preocupación por la seguridad derivó en la eliminación del mercado de productos efectivos para la pérdida de peso tales como fenfluramina y fentermina. Aunque la fentermina esta aún disponible como droga de prescripción en algunos países, la misma no esta avalada por el Real Colegio de Médicos a causa de los inadecuados datos sobre seguridad a largo plazo (20).
Definición de Eventos Adversos
Las efectos colaterales de las terapias con drogas son descritos utilizando los términos reacciones adversas de la droga (ADR) y/o eventos adversos (AE). Aunque las definiciones de estos conceptos son diferentes, nosotros utilizaremos eventos adversos como un término abarcativo y utilizaremos la definición de la Organización Mundial de la Salud (OMS) que define a los eventos adversos como “cualquier acontecimiento médico adverso en un paciente o en un sujeto de una investigación clínica, al cual se le administró un producto farmacéutico y que no necesariamente tiene que poseer una relación causal con este tratamiento” (24). De esta manera, un AE es cualquier signo o síntoma desfavorable o imprevisto asociado temporalmente con la utilización de un producto (droga o suplemento); no obstante el AE puede o no estar relacionado al producto en si mismo. Estas relaciones temporales pueden incluir 1) un AE que ocurre brevemente luego de la iniciación del uso del producto, 2) un AE que ocurre con la utilización del producto a largo plazo, y 3) un AE que ocurre luego de que la terapia con el producto haya finalizado (16). Por ejemplo, Haller y cols. (39) reportaron eventos adversos (i.e., temblores, palpitaciones, inquietud) a la hora y a las cinco horas de haberse administrado efedra, mientras que estudios caso han reportado AE severos (e.g., infarto de miocardio, accidente cerebro-vascular, psicosis, muerte) luego de días a años de utilización de efedra (38, 61, 64).
Hay dos categorías distintas de AE. La primera categoría, o eventos “tipo A”, son eventos que son predecibles por naturaleza, debido a que están relacionados con la dosis como una función de las acciones farmacológicas de los ingredientes activos (50). La susceptibilidad de los individuos a estos tipos de eventos puede ser dependiente de las diferencias en la disposición del individuo a la droga (cinética farmacológica) y/o a la respuesta del tejido a la droga/suplemento (dinámica farmacológica). Por ejemplo, la efedra es un agonista α y β adrenérgico y puede incrementar la frecuencia cardiaca. De esta manera, se puede establecer una relación directa entre la concentración plasmática y la frecuencia cardiaca, y la disminución de la dosis puede reducir la magnitud del incremento en la frecuencia cardiaca. Se han estudiado los efectos de la cantidad de dosis de efedrina/cafeína sobre el rendimiento en el ejercicio (8) y la perdida de peso (6, 56), sin embargo, no están disponibles estudios sobre verdadera tolerabilidad para hallar una dosis “óptima” de efedra/cafeína y como la misma se relaciona con la disminución de los AE, mientras aumenta la pérdida de peso.
La segunda categoría de AE, o eventos “tipo B”, es idiosincrásica, impredecible por naturaleza y frecuentemente parece no estar relacionada a la dosis o a la actividad farmacológica de la droga. Estos últimos eventos son relativamente raros (>1 en 10000), generalmente no mejoran con la reducción de la dosis, y son mas difíciles de diferenciar de los estados de enfermedad subyacentes. En la investigación farmacéutica, los eventos tipo B son los únicos detectados luego de que la droga halla estado disponible por varios años y halla sido utilizada por un gran número de pacientes. Por ejemplo, las anormalidades en las válvulas cardiacas, asociadas con la combinación de perdida de peso y Fen-Fen (fenfluramina y fentermina), que aparecieron años después de que se aprobara la comercialización de las drogas, podrían considerarse un evento tipo B. Para la efedra, un ejemplo de un evento idiosincrásico podría ser el caso reportado de una mujer de 32 años de edad que tuvo un parto prematuro y la subsecuente muerte de 4 neonatos, después de 4 años de consumo de 20 mg de efedra por día y un historial de adicción al cigarrillo (38). No pudo ni puede establecerse una relación de causa efecto directa con la efedra. Nuevamente, debido a la poca frecuencia y a la dificultad para determinar la causalidad en los eventos tipo B, nos enfocaremos mayormente en la discusión de los eventos tipo A.
Probando la Seguridad
La FDA (EE UU) y otras agencias mundiales de la salud tales como la Institución Canadiense de Protección de la Salud (HPB, Canadá), la Agencia de Regulación de Productos Medicinales y para el Cuidado de la Salud (MHRA, Reino Unido), y la Agencia Europea para la Evaluación de Productos Medicinales (EMEA, Europa) se han involucrado en la regulación potencial de los suplementos dietarios dado el hecho de que todavía no existe un sistema eficiente. Tres ejemplos de la implicación de la FDA son: 1) el establecimiento de un borrador de guía (i.e., un documento no implementado con una posible implementación futura) para productos botánicos (23); 2) en 1997, la FDA propuso (y retiró posteriormente) que la dosis diaria máxima de efedrina en un suplemento no debía ser mayor de 24 mg y que la dosis única no debía exceder los 8 mg (26); y 3) más recientemente en Diciembre de 2003 removió del mercado los productos de efedrina herbaria. De esta manera, es importante entender los objetivos de los cuerpos regulatorios con respecto a la seguridad. Los objetivos generales de una “revisión sobre seguridad” son (1) identificar AE importantes que estén causalmente relacionados al uso de la droga, (2) estimar la incidencia de estos eventos, e (3) identificar los factores que pueden predecir la ocurrencia de estos eventos (25); estos objetivos pueden cumplirse por medio de un diseño experimental apropiado.
En una revisión reciente, se concluyó que cuando se determina la eficacia de los suplementos para la mejora del rendimiento, es necesario enfocarse en tres puntos (62). Estos tres puntos pueden también aplicarse a los objetivos de una “revisión sobre la seguridad” (y la seguridad de los suplementos) e incluyen: 1) diseño del estudio, 2) factores asociados con la sustancia y 3) factores asociados a los sujetos. Las diferencias entre la valoración de la eficacia y valoración de la seguridad durante las pruebas clínicas características son 1) las hipótesis de seguridad para los AE característicos no están generalmente especificadas a priori, y 2) las pruebas clínicas no están comúnmente preparadas para evaluar las hipótesis con respecto a eventos de seguridad que no son frecuentes (19). Sin embargo, aunque no puedan alcanzarse diferencias estadísticas entre los grupos, ningún hallazgo con respeto a la seguridad debería ser ignorado.
DISEÑO DEL ESTUDIO
Identificación y Monitoreo de los AE
El primer objetivo de una revisión sobre seguridad es identificar los AE que están relacionados al uso del producto. Los estudios que intentan evaluar la seguridad de los suplementos dietarios deben primero identificar los AE relevantes o los marcadores biológicos apropiados (i.e., marcadores sustitutos) que pueden predecir los AE. Esto puede significar una variedad de mediciones fisiológicas, patológicas o anatómicas pensadas para relacionar algunos aspectos normales o patológicos de los procesos biológicos o psicológicos (51). Por ejemplo, los alcaloides de la efedra son compuestos que imitan las acciones del sistema simpático (41). Así, pueden afectar el sistema cardiovascular (CV) y al sistema nervioso central (SNC) y por lo tanto, los parámetros del CV (e.g., presión sanguínea y frecuencia cardiaca) y del SNC (e.g., evaluaciones psicométricas y evaluaciones de rendimiento) son mediciones apropiadas de AE. Los resultados de varias evaluaciones de laboratorio están influenciados por la dieta, el ambiente físico, la actividad física, el estado mental, el ritmo circadiano/biológico, e infecciones virales subclínicas (32). Por ello, los investigadores deben ser cuidadosos en la interpretación de los marcadores biológicos usados como mediciones sustitutas de los AE. Los parámetros cardiovasculares tales como la frecuencia cardiaca y la presión sanguínea pueden tener una gran variabilidad a lo largo del día, y es posible que la variabilidad normal en la frecuencia cardiaca y la presión sanguínea del sujeto pueda hacer que los cambios asociados con suplementos como la efedra sean difíciles de detectar. En numerosos estudios que han reportado los efectos de los suplementos de efedrina/efedra sobre el sistema cardiovascular, no se ha reportado la variabilidad de las mediciones (3, 4, 6, 3-15). Por ello es necesario establecer valores basales confiables, como así también un control con placebo, para ayudar a eliminar la variabilidad en la valoración clínica.
También es crucial una adecuada duración del estudio para la detección de AE. El tiempo de administración del suplemento es crítico, ya que algunos AE son mucho más comunes en la fase temprana de la terapia (48). No se sabe muy bien como esto se relaciona con la efedra, los reportes de la frecuencia de AE con el uso de efedrina son más comunes en las primeras 4 semanas de uso y disminuyen con el uso prolongado (3, 5, 15). Aunque algunos individuos utilicen efedra para la perdida de peso a corto plazo, otros la consumen durante largos períodos de tiempo para el tratamiento de la obesidad. Las directivas de la FDA para una evaluación segura de las terapias a largo plazo de enfermedades que no presentan una amenaza para la vida, afirman que la mayoría de los AE ocurren dentro de los primeros pocos meses del tratamiento con la droga y que el número de pacientes tratados durante 6 meses con dosis destinadas al uso clínico, debería ser adecuado para caracterizar el patrón de los AE a lo largo del tiempo (28). Sin embargo, la cohorte de sujetos expuestos debería ser lo suficientemente grande para observar si hay un incremento o una disminución de los AE a lo largo del tiempo, y para observar eventos que aparecen con un retraso en el tiempo con una frecuencia razonable (28). Es probable que los estudios que duran solo unas pocas semanas, con una muestra pequeña y más importante, con una valoración poco frecuente de los marcadores biológicos de los AE, no tienen la fortaleza necesaria para detectar o predecir la tasa de ocurrencia de AE.
Tamaño de la Muestra
El segundo objetivo de la revisión sobre la seguridad fue estimar la tasa de incidencia de los AE. La muestra de un estudio destinado a tratar puntos referentes a la seguridad de un suplemento puede necesitar un mayor número de sujetos que la muestra de un estudio destinado a tratar la eficacia (29). Los AE más comunes (e.g., inquietud, insomnio, frecuencia cardiaca incrementada, sequedad de la boca) ocurren con una frecuencia <1 en 100 individuos (67). Si un AE ocurre en 1 de cada 100 pacientes, se necesitaran 300 pacientes para detectar efectos adversos con el 95% de confianza (67). El punto referente al número de sujetos se complica además por los antecedentes generales de la tasa de incidencia. Debido a que los AE pueden estar relacionados a la droga, al estudio, o no estar relacionados con los mismos, son necesarios los grupos placebo para distinguir entre los AE relacionados al suplemento y los AE no relacionados al suplemento (i.e., tasa de incidencia de AE debido a la población versus la tasa de incidencia debida al suplemento). La Figura 1 ilustra el punto de la tasa de incidencia de AE en el grupo placebo comparada con un grupo experimental al que se le administró efedrina.
Lógicamente, las muestras más grandes proporcionan la mejor oportunidad de detectar y estimar la tasa de incidencia de los AE menos frecuentes. Las Fases I, II, y III de los estudios clínicos en el desarrollo de una droga tienen muestras que oscilan entre <100 a >1000 sujetos (Tabla 1). La fase III de las pruebas clínicas, debido al tamaño de la muestra, tendrían la mayor capacidad para estimar la tasa de incidencia de AE, pero, no es común en estudios con suplementos dietarios tener una muestra tan grande como la que tienen en la Fase III las pruebas clínicas. Estudios recientes sobre suplementos de efedra/cafeína y AE cardiovasculares reportaron hallazgos discordantes en cuanto a eventos cardiovasculares tipo A (13, 14, 45). Las diferencias en el tamaño de la muestra [30 (45) vs 67 (14) vs 167 (13)], en el control de los factores de confusión (consumo de cafeína, o medicamentos), y de los parámetros cardiovasculares estables (valoración continua vs. valoración en diferentes puntos en el tiempo) pueden explicar estas discordancias en los hallazgos. Boozer y cols. (14) reportaron el porcentaje de pacientes que manifestaron AE, los cuales fueron agrupados por sistemas corporales, luego de la ingestión de efedra/cafeína o placebo (Figura 1). Algunos AE fueron reportados con una frecuencia similar tanto en el grupo experimental como en el grupo control (sistema nervioso: ansiedad; sistema CV: palpitación; otros sistemas: dolor en el pecho), mientras que otros fueron bastante divergentes (sistema digestivo: otros; sistema nervioso: insomnio; sistema CV: general; otros sistemas: sequedad de boca). La frecuencia de AE del sistema nervioso reportada por los sujetos del grupo placebo fue 25% (ansiedad), 40% (insomnio), y 55% (otros). Estos datos ilustran otros puntos que deben considerarse cuando se diseña un estudio para detectar eventos adversos: 1) la importancia del grupo placebo para la comparación y 2) la necesidad de clasificar los AE en base a los sistemas corporales.
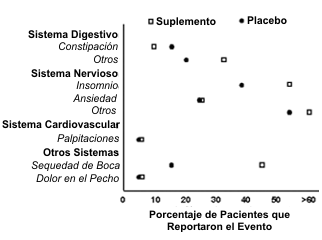
Figura 1. Porcentaje de pacientes que reportaron eventos adversos luego de
la ingestión de efedra/cafeína (63 mg/día de efedrina, 300 mg/día de cafeína)
o placebo durante 8 semanas en individuos con sobre peso. Datos resumidos de
la referencia 13.

Tabla 1. Objetivos globales de las pruebas con drogas pre comercialización
(fase I a III) y post comercialización (fase IV) con énfasis en la evaluación
de la seguridad en voluntarios saludables y en poblaciones de pacientes.
Resumido de Gad (32).
Una preocupación adicional es la falta de evaluación y seguimiento de los abandonos en los estudios de suplementación. Boozar y cols. (13) reportaron que 8 sujetos que recibían placebo y 7 sujetos que recibían el suplemento de efedra/cafeína abandonaron el estudio como resultado de AE. Hallazgos similares pueden observarse con agentes para la perdida de peso aprobados por la FDA tales como la sibutramina, donde en un estudio con este suplemento el abandono debido a AE fue mayor en el grupo placebo que en el grupo bajo tratamiento (66). Se puede obtener valiosa información de los sujetos que no completan el estudio, y por esta razón, estos individuos deben ser rastreados, evaluados cuando sea posible, e incluidos en los resultados del estudio. Los estudios más largos y con muestras más grandes con efedrina/cafeína y con efedra/cafeína herbal, suplementaron 141 sujetos durante 6 meses (3) y 167 sujetos durante 6 meses (13), respectivamente. Raramente se publican el número y las razones por las cuales los sujetos abandonaron los estudios con suplementación dietaria.
Debido a que los estudios de larga duración y con muestras de gran tamaño, son costosos y difíciles de manejar, la investigación sobre la seguridad de una droga comienza con lo que se denomina un primer estudio en humanos (FTIH). En estos estudios la evaluación de la tolerancia y la seguridad junto con la cinética farmacológica son los principales objetivos. Los estudios FITH son realizados bajo la guía de datos toxicológicos previos en animales, y requieren una serie de dosis (y placebo) únicas, con niveles ascendentes para tratar de determinar la máxima dosis tolerable (MDT). Estos tipos de estudio derivan un índice de eficacia/seguridad “ideal” que se determinara en las Fases II y III de las pruebas clínicas. Estos estudios implican un riguroso seguimiento de los sujetos (i.e., evaluaciones de sangre/orina, monitoreo Holter y exámenes físicos) y además el monitoreo subsiguiente del programa de dosaje (i.e., electrocardiograma de 12 derivaciones, signos vitales frecuentes, y evaluaciones químicas de sangre y orina); nuevamente, la seguridad es el interés principal. Aunque se utilizan una variedad de diseños para los estudios FTIH, un ejemplo se muestra en la Figura 2.
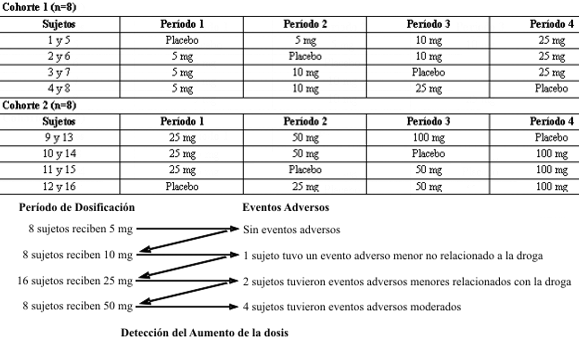
Figura 2. Ejemplo del procedimiento de aumento de la dosificación
(estudio FTIH) para la determinación de la máxima dosis tolerable.
Otros puntos metodológicos incluyen: valoración de la exposición a la droga, dosaje múltiple; y determinación de la causalidad. Las pruebas clínicas deberían además estudiar múltiples dosis del suplemento a corto plazo (la longitud del estudio depende comúnmente de la vida media del ingrediente activo) y valorar y documentar repetidamente cualquier evento adverso común posible; y como en los estudios FTIH, los sujetos son mantenidos de manera característica “en su casa” a lo largo de la duración del estudio. Un método para determinar la causalidad, relacionando los AE al producto y no a otras causas subyacentes, es a través del enfoque de la fase 4. La fase 4 incluye: 1) refuerzo positivo 2) refuerzo negativo 3) nuevo refuerzo positivo 4) nuevo refuerzo negativo. Aunque esta técnica puede ayudar a responder acerca de la cuestión de causalidad, durante las pruebas con drogas los refuerzos no son comúnmente realizados. Otro método utilizado para determinar la causalidad de los efectos adversos de una droga (o suplemento) en un paciente es la utilización de un cuestionario acerca de los AE y su relación con la exposición a la droga (57). Es común que el médico de un estudio sobre drogas determine la causalidad del evento adverso.
Factores Asociados con la Sustancia
El tercer objetivo de una revisión sobre la seguridad es identificar los factores que puedan predecir la ocurrencia de los AE. Esto puede incluir: pureza del producto, exposición al producto (cinética farmacológica), y factores asociados con los sujetos.
Pureza del Producto
Para los productos a base de hierbas, esto ofrece un nuevo desafío, ya que los ingredientes activos no se conocen y la calidad de los suplementos a base de hierbas pude ser ampliamente variable. En nuestro ejemplo de la efedra, la efedra herbaria es una mezcla compleja de alcaloides que incluye efedrina, pseudoefedrina, norefedrina, y metilefedrina. El contenido de efedrina de las plantas de efedra es dependiente de la especie, en donde crece la planta, el tipo condiciones en la cual crece, y el tiempo de cosecha (63). El contenido absoluto de alcaloides de efedrina y el índice de alcaloides de efedrina varía marcadamente, tanto dentro como entre partidas del mismo producto (35, 37). Gurley y cols. (37) reportaron que la desviación en el contenido total de alcaloides de efedrina en las afirmaciones de contenido era tan baja como 0%, a tan alta como 154%. La identificación y cuantificación de los elementos activos en mezclas complejas es necesaria para determinar la causalidad y relacionar la causalidad a la exposición a la droga. Claramente, esta alta variabilidad, tanto dentro de los productos como entre los productos, confunde los hallazgos de cualquier estudio que intente estudiar la asociación entre la ingestión de efedra y los AE. Los investigadores deberían proporcionar análisis independientes del contenido de los suplementos en sus artículos publicados, pero actualmente esta práctica no es común. En lugar de esto, los investigadores podrían obtener una verificación escrita de los fabricantes del suplemento indicando el nivel de pureza del producto y si siguieron o no procedimientos de fabricación apropiados. Debería ser destacado, que se han propuesto lineamientos para las Practicas Apropiadas de Fabricación para los suplementos dietarios (GMP) (2).
Exposición al Producto
“La información sobre la respuesta a la exposición al producto es el centro de cualquier determinación sobre la seguridad y la efectividad de las drogas. Esto es, solo puede determinarse que una droga es segura y efectiva cuando se conoce la relación de efectos benéficos y adversos ante una exposición definida” (28). Exposición se refiere a poner la droga dentro del cuerpo (i.e., la dosis), a la vez que pueden utilizarse varias mediciones de concentraciones agudas o integradas en plasma o en otros fluidos biológicos, para caracterizar la exposición (e.g., concentraciones máximas o CMAX, concentraciones promedio, área bajo la curva o AUC) (27).
El entendimiento de la cinética farmacológica del suplemento (i.e., la exposición) ayuda a predecir el potencial para AE. A partir de la perspectiva de la droga, los datos obtenidos en animales proporcionan pautas acerca de las concentraciones sanguíneas asociadas con AE, o más precisamente, las concentraciones que no están asociadas con AE. Debido a que realmente no se conoce la calidad de los productos a base de hierbas, se necesitan evaluaciones independientes para asegurar la cantidad en las dosis. Las muestras sanguíneas son necesarias para determinar la exposición a la droga y para relacionar como se relaciona la exposición a los EA. En general, los individuos que eliminan un componente lentamente (i.e., una función renal lenta o una acción lenta de las enzimas metabólicas) pueden tener una mayor exposición sistémica a un ingrediente activo y por lo tanto derivar en un AE; este tipo de información específica del paciente no se conocería sin la cuantificación de la exposición. Además, la medición clínica de la variable independiente para valorar los EA (i.e., el resultado de la dinámica farmacológica) podría estar retrasada o ser persistente con respecto a los niveles sanguíneos tal como se sugirió anteriormente. Este tipo de efectos resulta de una relación respuesta-exposición con una considerable histéresis (efecto bucle). Aún en este caso, las relaciones de respuesta-exposición pueden ser informativas. La efedra, por ejemplo, sigue una histéresis en el sentido de las agujas del reloj para los efectos sobre la presión sanguínea (10, 39). Una histéresis en sentido de las agujas del reloj es indicativa de tolerancia aguda (10, 54), similar a los efectos observados con la cocaína, un compuesto farmacológicamente relacionado a la efedrina (70).
Preparados a Base de Hierbas vs. Preparados Sintéticos
Los ingredientes activos tienen efectos similares sin considerar si son preparados sintéticos o preparados a base de hierbas, siempre y cuando la estructura molecular sea idéntica. Sin embargo, dado que ciertos productos herbarios/dietarios contienen otros moduladores potenciales, los efectos farmacológicos pueden aumentarse o disminuirse. Por ejemplo, desde el punto de vista farmacodinámico (i.e., relación dosis – respuesta) la efedrina provocaría la misma respuesta farmacológica (e.g., termogénica o cardiotóxica) sin considerar la fuente de la efedrina (i.e., derivada de la planta o sintética) a menos que otra substancia (e.g., cafeína, aspirina, pseudoefedrina) este también presente. De este modo, mientras las concentraciones sean similares en el sitio de acción, la extensión de los resultados farmacológicos/toxicológicos asume que no están presentes otras moléculas de confusión del producto. Por ejemplo, Berlin y cols. (10) hallaron incrementos pico en la presión sanguínea de ~15% luego de una dosis de 50 mg; mientras que Haller y cols. (39) hallaron aproximadamente los mismos incrementos pico en la presión sanguínea (~15%) con una dosis mas pequeña de efedrina (17.3 mg), pero esta fue dada en la forma herbaria junto con cafeína (175 mg de cafeína). Esto sugiere que la combinación de los productos, observada de manera característica, en los productos de la efedra puede potenciar los efectos cardiovasculares más allá de los producidos solamente por la efedrina.
La similitud en la exposición a la droga para diferentes productos es la base para el principio de la bioequivalencia de los productos farmacéuticos o para probar que un producto puede cambiarse por otro producto sin perdida de la eficacia. Gurley y cols. (369) hallaron que casi no había diferencias en la concentración plasmática y el perfil temporal entre el hidrocloruro de efedrina sintética (HCl, 25 mg) y 3 productos a base de efedra herbaria con un contenido equivalente de efedrina. Asumiendo que esto productos son bioequivalentes, lo cual es razonable, en base a los errores estándar de el área bajo la curva y la CMAX en este estudio, y siendo la exposición del cuerpo a la efedrina similar, entonces las respuestas farmacológicas/toxicológicas deberían ser también similares. De esta manera, el perfil de los efectos colaterales para la efedrina, un producto OTC aprobado por la FDA, debería ser similar al de los productos de la efedra. Debe resaltarse que estos son lo efectos de la efedrina por si sola y no de otros alcaloides de la efedra y cafeína los cuales están comúnmente incluidos en los productos de la efedra.
Factores Asociados a los Sujetos
En un estudio sobre la seguridad, la caracterización de la población que compone el estudio y el monitoreo apropiado de los sujetos es de extrema importancia. Las poblaciones que utilizan suplementos dietarios están en un rango que va desde jóvenes, adultos aparentemente saludables, a ancianos a los cuales se les prescriben múltiples medicamentos, a personas obesas con comorbilidad múltiple. Las características de los sujetos pueden usarse para identificar los factores que potencialmente podrían predisponer al individuo a un AE (e.g., edad, sexo, raza, enfermedades comorbidas). La descripción de los sujetos debería incluir el sexo, el tamaño corporal (y posiblemente la composición corporal), la etnia, condiciones médicas pre existentes, el uso de medicamentos, y la exposición a los componentes que están siendo evaluados. La actividad física previa o el nivel de aptitud física, así como también la dieta y las comidas previas pueden también ser informativas.
El uso concomitante de la droga más allá de la dosis establecida o de drogas ilegales puede tener un efecto significativo sobre los eventos adversos. Para las drogas aprobadas, la interacción con otras drogas explica el 25% de los reportes de eventos adversos (48) y son estas interacciones (i.e., droga-suplementos) que pueden ser parcialmente responsables de los EA observados en la literatura sobre suplementos dietarios. Un ejemplo de la interacción droga-efedra es un estudio de caso en donde se reporto que un hombre relativamente sano que usaba efedrina (Thermadrene®) sufrió de un accidente cerebro-vascular isquémico (44). Este sujeto fumó un paquete de cigarrillos por día durante 12 años y estaba tomando bupropion, un neurotransmisor inhibidor usado para dejar de fumar. Los autores de este reporte concluyeron que la efedra fue responsable del evento adverso y desestimaron la posible interacción con el bupropion. El bupropion interactúa con otras drogas entre las que se incluyen la cocaína, las anfetaminas y otros estimulantes, drogas con mecanismos de acción similar al de la efedrina. Es más probable que la interacción entre el bupropion y la efedra sea causa de AE que la efedra en si misma. Esto demuestra el punto que establece que es necesario señalar y reportar los medicamentos que son consumidos concomitantemente en un estudio sobre la seguridad.
Las evaluaciones clínicas tempranas para las drogas (i.e., la fase I de las pruebas clínicas de una droga en desarrollo) generalmente usan voluntarios saludables para reducir la posibilidad de efectos colaterales o anormalidades que podrían ser causadas por una enfermedad subyacente u otros medicamentos. A medida que se define el perfil de los efectos colaterales, la evaluación se mueve hacia pruebas con poblaciones de pacientes (Fase II y III). En el caso de suplementos dietarios, los estudios iniciales sobre la seguridad deberían realizarse en voluntarios saludables que estén libres de una patología/enfermedad concomitante, uso o abuso de drogas o alcohol y de cualquier otro factor que probablemente pueda causar confusión en la interpretación de los resultados. Es probable que muchas personas que sufren de enfermedades estén usando suplementos dietarios a pesar de la falta de estudios que documenten la seguridad para utilizar el suplemento en esa población con una enfermedad específica. Estudios recientes han examinado la eficacia y los efectos colaterales de la efedra en individuos “saludables” aunque con sobrepeso (13, 14).
La exposición previa a la efedra y a la efedra/cafeína puede causar
confusión en los estudios sobre efectos adversos ya que se observa una
tolerancia (o sensibilidad) tanto para la efedrina como para la cafeína (30,
69). Así mismo, un sujeto que es tratado por primera vez con efedrina o
cafeína puede tener un efecto farmacológico mayor que en un sujeto que esta
acostumbrado al consumo, resultando en un AE donde un consumidor frecuente que
ha creado una tolerancia puede no demostrar ningún efecto colateral. Por ello,
cuando se monitorea a los sujetos los investigadores deberían caracterizar a
los mismos como consumidores, consumidores excesivos, no consumidores, y
consumidores sensibles. Mientras los beneficios de estudiar a un grupo por
sobre otro no son claros, parece prudente incluir a todos los grupos en el
mismo estudio. Recientes estudios sobre efedra excluyeron tanto a los
consumidores de cafeína (> 500 mg/día) (13, 14) como a los sujetos sensibles a
la cafeína (45). Además, la obesidad, la resistencia a la insulina y el estrés
contribuyen a la activación simpática crónica (21) y por ello la adición de un
componente que imite los efectos del sistema simpático como la efedra podría
provocar eventos adversos a una tasa más frecuente que en poblaciones normales
y saludables.
No es necesario un diseño de investigación ideal o una lista de parámetros
para valorar la seguridad en la investigación sobre los suplementos dietarios,
pero los diseños de investigación que siguen el tipo de diseño FTIH podrían
proporcionar información valiosa sobre la seguridad y tolerancia de un
suplemento dietario. En la Tabla 2 se presentan algunas variables que son
críticas para medir y reportar en los estudios de esta naturaleza. Algunas
variables tales como edad, sexo y peso corporal son comúnmente reportadas,
pero otros tales como la frecuencia de AE en relación a la dosis o la causa de
abandono de los sujetos, son reportadas con mucha menos frecuencia.
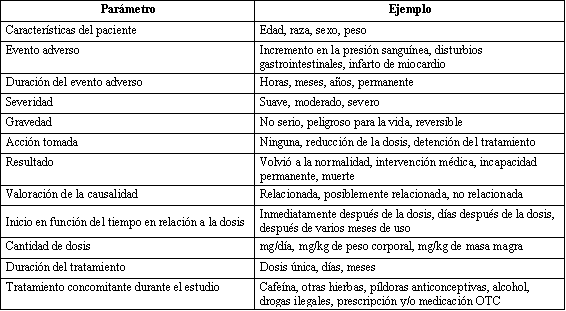
Tabla 2. Parámetros sugeridos para ser valorados en los estudios que
evalúan la seguridad de los suplementos dietarios.
DETECTANDO EVENTOS ADVERSOS EXTRAÑOS
Las pruebas con drogas antes de su comercialización (fase I a la III) son usadas para evaluar la seguridad en voluntarios saludables y en poblaciones de pacientes (Tabla 1). Sin embargo, estas pruebas clínicas no tienen la fortaleza suficiente para detectar eventos adversos más raros y a veces más serios, como los tipo B . A pesar del tiempo, dinero y de los esfuerzos puestos en la evaluación, aún se reportan AE después de la comercialización del producto (fase IV, posterior a la aprobación de la FDA) y en algunos casos resulta en que una amplia variedad de drogas son quitadas del mercado [e.g., Pondimin® (fenfluramina) y Redux® (dexfenfluramina) por anormalidades en las válvulas cardiacas; Saldane® (terfenadina) por anormalidades fatales en el ritmo cardiaco; Renzulin® (troglitazona) por toxicidad hepática, fenilpropanolamina OTC por accidente cerebrovascular hemorrágico; Baycol® (cerivastatina) por rabdomilosis]. Los sistemas actualmente disponibles para estudiar AE extraños asociados con los suplementos dietarios sobre un nivel poblacional luego de que un producto halla sido vendido durante varios años es el sistema Medwatch de Estados Unidos (www.fda.gov/medwatch) o el sistema “Yellow Card” del Reino Unido. Dos estudios han utilizado las bases de datos del gobierno de los Estados Unidos para reportar efectos adversos con el uso de la efedra (38, 64). Utilizando el sistema Medwatch, Haller y Benowitz (38) revisaron 140 reportes de EA presentados a la FDA (1997 a 1999) y aplicaron un sistema de clasificación para la valoración de la causalidad. Treintiuno porciento de los casos fueron considerados definitiva o probablemente relacionados al uso de suplementos que contenían alcaloides de la efedra, y 31 porciento fueron considerados como posiblemente relacionados. Los EA incluyeron hipertensión (17 reportes), palpitaciones, taquicardia o ambos (13) accidente cerebro-vascular (10), y convulsiones (7). Diez eventos resultaron en la muerte del individuo, y 13 eventos resultaron en discapacidad permanente. Samenuk y cols. (64) revisaron la base de datos del ahora desaparecido Sistema de Monitoreo de Reacciones Adversas de la FDA para eventos relacionados a la efedra. De 926 casos de AE reportados a la FDA (1995 a 1997), el uso de ma huang en 37 pacientes fue temporalmente relacionado al accidente cerebre-vascular (16), infarto de miocardio (10) o muerte súbita (11). Se reportó que el uso de ma huang estaba dentro de las dosis recomendadas por los fabricantes en 36 de los 37 pacientes. Los autores concluyeron que el uso de ma huang se relaciona temporalmente al accidente cerebre-vascular; al infarto de miocardio, y la muerte súbita; que las enfermedades cardiacas o vasculares subyacentes no son un pre requisito para los eventos adversos relacionados al ma huang; y que los eventos adversos cardiovasculares asociados al ma huang no se limitan a las dosis masivas (64).
Estos estudios proporcionan fuerte evidencia circunstancial que indica que los alcaloides de la efedra pueden estar asociados a complicaciones médicas serias, pero no establecen una relación causal. Los eventos adversos reportados por Haller y Benowitz (38), por Samenuk y cols. (64) y por Shekelle y cols. (65) son consistentes con los AE esperados para drogas que imitan los efectos del sistema simpático, respaldando la validez de sus hallazgos. Sin embargo, los factores de confusión tales como una enfermedad subyacente o la interacción con otras drogas no fueron controlados lo cual debilita las conclusiones que se pueden realizar a partir de estudios de esta naturaleza. Además, debido a que estos reportes de AE son comunicados voluntariamente, puede ocurrir que se produzca una subestimación en el número de reportes, y por ello no es posible estimar el número de AE asociados con la ingestión de efedra en base a la población. El Centro para la Seguridad de los Alimentos y Nutrición Aplicada de la FDA está actualmente desarrollando un sistema para rastrear y analizar eventos adversos asociados con los suplementos dietarios.
REPORTANDO LOS DATOS
Más comúnmente, los AE producidos por suplementos, son reportes de caso, y para las drogas los AE más serios son señalados en principio mediante este mecanismo. El estudio de caso puede servir para dos propósitos: 1) los estudios de caso pueden servir como una herramienta de monitoreo para investigaciones clínicas futuras, y 2) pueden reportar los eventos inusuales o raros que ocurren con el uso a largo plazo. Sin embargo, los reportes de caso están influenciados por la tasa a la cual ocurren los AE (i.e., frecuencia), por la relación temporal con respecto a la exposición al suplemento (i.e., si el AE ocurrió durante la utilización o luego de haber dejado de tomar el suplemento), y por el estilo de vida subyacente (i.e., tabaquismo, consumo de alcohol), y de enfermedad (16).
Como se mencionó anteriormente, los efectos colaterales de los suplementos, como en las drogas tradicionales, pueden separarse en eventos raros y comunes. En los eventos raros, cada evento es importante y respalda la necesidad de los estudios de caso. En estos eventos, los índices de personas expuestas al suplemento se vuelven muy útiles. Por ejemplo, dos estudios de Boozer y cols. (13, 14) reportaron efectos colaterales en estudios sobre perdida de peso con consumo de la efedra/cafeína, en un total de 230 individuos saludables, pero con sobrepeso. Estos estudios tuvieron una duración de 8 semanas a 6 meses y el grupo que consumió el suplemento tuvo una mayor incidencia de efectos colaterales (69%) que el grupo control con administración de placebo (50%). Un punto importante, los sujetos podrían haber reportado más de un efecto colateral, el cual no hubiera sido detectable proporcionando solo los números de la frecuencia. Por esta razón, la utilización de un índice de incidencia bruto, tal como (número de sujetos con AE/ número de sujetos que recibieron el suplemento), tomaría en cuenta a los sujetos con múltiples AE; aunque este cálculo no tiene en cuenta la exposición ni la severidad y duración de los AE (19). Por ejemplo, la Figura 1 describe la frecuencia de AE en base a la población total del estudio y no ilustra cuantos sujetos reportaron AE. Sin embargo, la Tabla 3 es una tabla de contingencia que discrimina los efectos adversos en base al número de sujetos que reportaron AE, el número de AE, y el número total de sujetos a partir de los cuales pueden calcularse los datos referentes a la frecuencia.

Tabla 4. Eventos adversos reportados durante la comparación de la
eficacia entre la dexfenfluramina y la efedrina/cafeína (E+C) en individuos
con sobrepeso. Los datos están resumidos de Breum y cols (15).
DISCUSION
La falta de una adecuado conocimiento de la información sobre los suplementos dietarios por el público y por los practicantes de la medicina o el mal uso de los suplementos por parte de los consumidores, son probablemente los principales factores que contribuyen en la aparición de AE. La confusión que circunda a los suplementos dietarios y la falta de datos en lo concerniente a la seguridad de muchos suplementos no parece haber disuadido a los consumidores, ya que las ventas de suplementos se ha incrementado en los Estados Unidos desde 9.8 a 14.7 billones de dólares (1995 a 1999) y se proyecta una venta que alcanzará los 18.5 billones de dólares en el 2002; y se ha estimado que hay 12 millones de consumidores de suplementos en los Estados Unidos (1). Estados Unidos ha observado el mayor crecimiento en las ventas a los largo de la década pasada, pero las ventas ha nivel mundial también se han incrementado (18). Así mismo, la Dirección de Suplementos Dietarios (ODS) del Instituto Nacional de Salud de Estados Unidos (NIH) lanzó en 1998 una campaña en donde dos de sus objetivos fueron “mejorar la metodología científica relacionada al estudio de los suplementos dietarios” e “informar y educar a los científicos, sujetos relacionados con el cuidado de la salud y al publico en general acerca de los riesgos y beneficios de los suplementos dietarios” (59). Las consideraciones metodológicas concernientes a la eficacia han sido establecidas en otros artículos (40, 62), sin embargo, algunos de los puntos respecto de la seguridad no han sido adecuadamente establecidos y pueden estar resumidos en la tabla 4.

Tabla 5. Resumen de los puntos principales en la determinación de eventos
adversos asociados a los suplementos dietarios.
En medio del frenesí mediático que desafía la seguridad de los suplementos dietarios, en Estados Unidos la población general desea tener una mayor certeza respecto a lo que concierne a los asuntos de seguridad de los suplementos, tal como la FDA proporciona para la prescripción y las drogas OTC (12, 49, 52). Sin embargo, la intervención del gobierno promueve el debate entre aquellos que quieren proteger un derecho individual de autotratamiento, y aquellos que priorizan la seguridad pública. La desconfianza entre los investigadores, el gobierno y los fabricantes de suplementos ha hecho difícil el desarrollo de un método sistemático para la evaluación de los suplementos dietarios. En un comentario reciente (49), C. Everett Koop, antiguo Cirujano General de Estados Unidos, afirmó “... la industria de productos naturales ha mantenido distancia de la investigación medica y de la práctica médica clínica, enfocándose en cambio en la ventaja comercial a corto plazo derivada de mantener a los productos a base de hierbas y remedios nutricionales exentos de cualquier revisión por parte de la FDA...”. Además, la desconfianza de los investigadores por la investigación sobre suplementos dietarios patrocinada por la industria, ha reducido la credibilidad de los estudios patrocinados por la industria (40, 71). Debido a esta desconfianza, se sugiere que la comunidad científica que investiga en nutrición sea responsable de evaluar el impacto a largo plazo de los suplementos dietarios (58).
La seguridad es un término relativo más que absoluto que esta basado en el índice riesgo beneficio y su valoración necesita ser establecida para demostrar que los beneficios de un suplemento pesan claramente más que los riesgos. Los factores tales como el metabolismo, genética, características demográficas de los consumidores (e.g., edad, sexo, raza), interacciones con medicamentos concomitantes, o estados fisiológicos y fisiopatológicos (e.g., enfermedades renales o hepáticas) deben ser estipuladas, ya que estos factores pueden ser evaluados y cuantificados y deben servir como la base fundamental de la valoración de los AE producidos por los suplementos dietarios. Una valoración eficiente, y el monitoreo y verificación de los AE puede derivar en un mejor entendimiento de los factores que tienen impacto en el desarrollo de los AE. La determinación de la seguridad y de la eficacia debe estar disponible de manera tal que los consumidores puedan elegir en base a la información sobre la utilización de los suplementos. Es necesario que esta sección proporcione suficientes datos e interpretaciones acerca de investigaciones anteriores para identificar claramente la necesidad de realizar su estudio. Sin embargo, la misma no debería ser tan larga. Enfocando el contenido para que sea capaz de contestar varias preguntas importantes.
Agradecimientos
Los autores quisieran agradecer a la Dra. Priscilla M. Clarkson y al Dr. Frank Hoke por su energía en la preparación de este manuscrito. A. M. Persky tiene una Beca en Farmacocinética/Farmacodinámica Clínica otorgada por la Universidad de North Carolina en colaboración con GlaxoSmithKline.
Dirección para correspondencia
Persky AM, Ph.D., Division of Drug Delivery and Disposition, School of Pharmacy, CB 7360 Kerr Hall, University of North Carolina at Chapel Hill, Chapel Hill, N.C. 27599-7360, Teléfono: (919) 966-7144, Fax : (919) 966-0197, correo electrónico: apersky@nc.rr.com
Referencias
1. No Disponible (2002). Dietary supplement sales are slowing. Research Alert. 20:10,
2. No Disponible (2003). Federal Register. pp. 12157-12263
3. Astrup, A., L. Breum, S. Toubro, P. Hein, and F. Quaade (1992). The effect and safety of an ephedrine/caffeine compound compared to ephedrine, caffeine and placebo in obese subjects on an energy restricted diet. A double blind trial. Int J Obes Relat Metab Disord; 16:269-277
4. Astrup, A., B. Buemann, N. J. Christensen, S. Toubro, G. Thorbek, O. J. Victor, and F. Quaade (1992). The effect of ephedrine/caffeine mixture on energy expenditure and body composition in obese women. Metabolism; 41:686-688
5. Astrup, A. and S. Toubro (1993). Thermogenic, metabolic, and cardiovascular responses to ephedrine and caffeine in man. Int J Obes Relat Metab Disord; 17 Suppl 1:S41-43
6. Astrup, A., S. Toubro, S. Cannon, P. Hein, and J. Madsen (1991). Thermogenic synergism between ephedrine and caffeine in healthy volunteers: a double-blind, placebo-controlled study. Metabolism; 40:323-329
7. Barnes, J (2003). Quality, efficacy and safety of complementary medicines: fashions, facts and the future. Part I. Regulation and quality. Br J Clin Pharmacol; 55:226-233
8. Bell, D. G., I. Jacobs, T. M. McLellan, and J. Zamecnik (2000). Reducing the dose of combined caffeine and ephedrine preserves the ergogenic effect. Aviat Space Environ Med; 71:415-419
9. Benzi, G. and A. Ceci (1997). Herbal medicines in European regulation. Pharmacol Res; 35:355-36
10. Berlin, I., D. Warot, G. Aymard, E. Acquaviva, M. Legrand, B. Labarthe, I. Peyron, B. Diquet, and P. Lechat (2001). Pharmacodynamics and pharmacokinetics of single nasal (5 mg and 10 mg) and oral (50 mg) doses of ephedrine in healthy subjects. Eur J Clin Pharmacol; 57:447-455
11. Blanck, H. M., L. K. Khan, and M. K. Serdula (2001). Use of nonprescription weight loss products: results from a multistate survey. JAMA; 286:930-935
12. Blendon, R. J., C. M. DesRoches, J. M. Benson, M. Brodie, and D. E. Altman (2001). Americans views on the use and regulation of dietary supplements. Arch Intern Med; 161:805-810
13. Boozer, C. N., P. A. Daly, P. Homel, J. L. Solomon, D. Blanchard, J. A. Nasser, R. Strauss, and T. Meredith (2002). Herbal ephedra/caffeine for weight loss: a 6-month randomized safety and efficacy trial. Int J Obes Relat Metab Disord; 26:593-604
14. Boozer, C. N., J. A. Nasser, S. B. Heymsfield, V. Wang, G. Chen, and J. L. Solomon (2001). An herbal supplement containing Ma Huang-Guarana for weight loss: a randomized, double-blind trial. Int J Obes Relat Metab Disord; 25:316-324
15. Breum, L., J. K. Pedersen, F. Ahlstrom, and J. Frimodt-Moller (1994). Comparison of an ephedrine/caffeine combination and dexfenfluramine in the treatment of obesity. A double-blind multi-centre trial in general practice. Int J Obes Relat Metab Disord; 18:99-103
16. Brewer, T. and G. A. Colditz (1999). Postmarketing surveillance and adverse drug reactions: current perspectives and future needs. Jama; 281:824-829
17. Buemann, B., P. Marckmann, N. J. Christensen, and A. Astrup (1994). The effect of ephedrine plus caffeine on plasma lipids and lipoproteins during a 4.2 MJ/day diet. Int J Obes Relat Metab Disord; 18:329-332
18. Calixto, J. B (2000). Efficacy, safety, quality control, marketing and regulatory guidelines for herbal medicines (phytotherapeutic agents). Braz J Med Biol Res; 33:179-189
19. Chow, S.-C. and J.-p. Liu (1998). Design and analysis of clinical trials: concept and methodologies. New York: Wiley, XI, 649
20. Collins, P. and G. Williams (2001). Drug treatment of obesity: from past failures to future successes?. Br J Clin Pharmacol; 51:13-25
21. Curtis, B. M. and J. H. O'Keefe, Jr (2002). Autonomic tone as a cardiovascular risk factor: the dangers of chronic fight or flight. Mayo Clin Proc; 77:45-54
22. Dulloo, A. G. and D. S. Miller (1986). The thermogenic properties of ephedrine/methylxanthine mixtures: human studies. Int J Obes; 10:467-481
23. FDA (2000). Botanical drug products: Draft guidance. 23. FDA. Botanical drug products: Draft guidance. Rockville: Food and Drug Administration
24. FDA (1995). Clinical safety data management: Definitions and standards for expedited reporting. Rockville: Food and Drug Administration, Publication ICH-E2A
25. FDA (1996). Conducting a Clinical Safety Review of a New Product Application and Preparing a Report on the Review: Draft guidance. Rockville: Food and Drug Administration. Publication ICH-E9
26. FDA (1997). Dietary supplements containing ephedrine alkaloids. Available at: http://vm.cfsan.fda.gov/~lrd/fr97064a.html Accessed May 5
27. FDA (2002). Exposure-response relationships: Study design, data analysis, and regulatory applications: Draft guidance. Rockville: Food and Drug Administration
28. FDA (1995). The extent of population exposure to assess clinical safety: for drugs intended for long-term treatment of non-life-threatening conditions. Rockville: Food and Drug Administration, Publication ICH-E1A
29. FDA (1998). Statistical methods for clinical trials. Rockville: Food and Drug Administration, Publication ICH-E9
30. Fredholm, B. B., K. Battig, J. Holmen, A. Nehlig, and E. E. Zvartau (1999). Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol Rev; 51:83-133
31. Fugh-Berman, A (1997). Clinical trials of herbs. Prim Care; 24:889-903
32. Gad, S. C (2002). Drug safety evaluation. New York: Wiley-Interscience, 1007
33. Green, G. A., F. D. Uryasz, T. A. Petr, and C. D (2001). Bray. NCAA study of substance use and abuse habits of college student- athletes. Clin J Sport Med; 11:51-56
34. Gurley, B (2000). Extract versus herb: effect of formulation on the absorption rate of botanical ephedrine from dietary supplements containing Ephedra (ma huang) (commentary). Ther Drug Monit; 22:497
35. Gurley, B. J., S. F. Gardner, and M. A. Hubbard (2000). Content versus label claims in ephedra-containing dietary supplements. Am J Health Syst Pharm; 57:963-969
36. Gurley, B. J., S. F. Gardner, L. M. White, and P. L. Wang (1998). Ephedrine pharmacokinetics after the ingestion of nutritional supplements containing Ephedra sinica (ma huang). Ther Drug Monit; 20:439-445
37. Gurley, B. J., P. Wang, and S. F. Gardner (1998). Ephedrine-type alkaloid content of nutritional supplements containing Ephedra sinica (Ma-huang) as determined by high performance liquid chromatography. J Pharm Sci; 87:1547-1553
38. Haller, C. A. and N. L. Benowitz (2000). Adverse cardiovascular and central nervous system events associated with dietary supplements containing ephedra alkaloids. N Engl J Med; 343:1833-1838
39. Haller, C. A., P. Jacob, and N. L. Benowitz (2002). Pharmacology of ephedra alkaloids and caffeine after single-dose dietary supplement use. Clin Pharmacol Ther; 71:421-432
40. Heyland, D. K (2001). In search of the magic nutraceutical: problems with current approaches. J Nutr; 131:2591S-2595S
41. Hoffman, B. B. and R. J. Lefkowitz (1996). Catecholamines, sympathomimetic drugs, and adrenergic receptor antagonists. In: Hardman, Limbird, Molinoff, Ruddon, and Gilman (Eds.) Goodman & Gilman s The Pharm. Basis of Therapeutics. N. Y.: McGraw-Hill, pp. 199-248
42. Huang, S. M., S. D. Hall, P. Watkins, L. A. Love, C. Serabjit-Singh, J. M. Betz, F. A. Hoffman, P. Honig, P. M. Coates, J. Bull, S. T. Chen, G. L. Kearns, and M. D. Murray (2004). Drug interactions with herbal products and grapefruit juice: a conference report. Clin Pharmacol Ther; 75:1-12
43. Hutchinson, T. A. and D. R. Shahan (2002). DRUGDEX System Greenwood Village. CO: MICROMEDIX
44. Kaberi-Otarod, J., R. Conetta, K. K. Kundo, and A. Farkash (2002). Ischemic stroke in a user of Thermadrene: a case study in alternative medicine. Clin Pharmacol Ther; 72:343-346
45. Kalman, D., T. Incledon, I. Gaunaurd, H. Schwartz, and D. Krieger (2002). An acute clinical trial evaluating the cardiovascular effects of an herbal ephedra-caffeine weight loss product in healthy overweight adults. Int J Obes Relat Metab Disord; 26:1363-1366
46. Kanayama, G., A. J. Gruber, H. G. Pope, Jr., J. J. Borowiecki, and J. I. Hudson (2001). Over-the-counter drug use in gymnasiums: an underrecognized substance abuse problem?. Psychother Psychosom; 70:137-140
47. Keller, K (1996). Herbal medicinal products in Germany and Europe: Experiences with national and European Assessment. Drug Inform J; 30:933-948
48. Kirby, D. S (1993). Adverse drug events in clinical trials. In: G. Sogliero-Gilbert (Ed.) Drug safety assessment in clinical trials. New York: Dekker, pp. 25-38
49. Koop, C. E (2002). The future of medicine. Science; 295:233
50. Lamy, P. P (1990). Adverse drug effects. Clin Geriatr Med; 6:293-307
51. Lesko, L. J. and A. J. Atkinson, Jr (2001). Use of biomarkers and surrogate endpoints in drug development and regulatory decision making: criteria, validation, strategies. Annu Rev Pharmacol Toxicol; 41:347-366
52. Lindsay, B. D (2002). Are serious adverse cardiovascular events an unintended consequence of the Dietary Supplement Health and Education Act of 1994?. Mayo Clin Proc; 77:7-9
53. Mahady, G. B (2001). Global harmonization of herbal health claims. J Nutr; 131:1120S-1123S
54. Meibohm, B. and H. Derendorf (1997). Basic concepts of pharmacokinetic/pharmacodynamic (PK/PD) modeling. Int J Clin Pharmacol Ther; 35:401-413
55. Miller, C. A (1998). How safe are herbs?. Geriatr Nurs; 19:163-164
56. Morgan, J. B., D. A. York, A. Wasilewska, and J. Portman (1982). A study of the thermic responses to a meal and to a sympathomimetic drug (ephedrine) in relation to energy balance in man. Br J Nutr; 47:21-32
57. Naranjo, C. A., U. Busto, E. M. Sellers, P. Sandor, I. Ruiz, E. A. Roberts, E. Janecek, C. Domecq, and D. J. Greenblatt (1981). A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther; 30:239-245
58. Nesheim, M. C (1998). Dietary supplements. Nutrition; 14:729-730
59. NIH (1998). Merging quality science with supplement research a strategic plan for the Office of Dietary Supplements. Bethesda, MD: National Institutes of Health
60. PhRMA (2002). PhRMA Industry Profile. Washington DC: Pharmaceutical Research and Manufacturers of America
61. Rawson, E. S. and P. M. Clarkson (2002). Ephedrine as an Ergogenic Aid. In: M. S. Bahrke and C. E. Yesalis (Eds.) Performance-Enhancing Substances in Sport and Exercise. Champaign, IL: Human Kinetics, pp. 289-298
62. Reents, S (2002). Determining the efficacy of performance-enhancing substances. In: M. S. Bahrke and C. E. Yesalis (Eds.) Performance-enhancing substances in sport and exercise. Champaign: Human Kinetics, pp. 21-32
63. Sagara, K., T. Oshima, and T. Misaki (1983). A simultaneous determination of norephedrine, pseudoephedrine, ephedrine and methylephedrine in Ephedrae Herba and oriental pharmaceutical preparations by ion-pair high-performance liquid chromatography. Chem Pharm Bull (Tokyo); 31:2359-2365
64. Samenuk, D., M. S. Link, M. K. Homoud, R. Contreras, T. C. Theohardes, P. J. Wang, and N. A. Estes (2002). 3rd. Adverse cardiovascular events temporally associated with ma huang, an herbal source of ephedrine. Mayo Clin Proc; 77:12-16
65. Shekelle, P. G., M. L. Hardy, S. C. Morton, M. Maglione, W. A. Mojica, M. J. Suttorp, S. L. Rhodes, L. Jungvig, and J. Gagne (2003). Efficacy and safety of ephedra and ephedrine for weight loss and athletic performance: a meta-analysis. Jama; 289:1537-1545
66. Smith, I. G. and M. A. Goulder (2001). Randomized placebo-controlled trial of long-term treatment with sibutramine in mild to moderate obesity. J Fam Pract; 50:505-512
67. Stein, C. M (2002). Are herbal products dietary supplements or drugs?. An important question for public safety Clin Pharmacol Ther; 71:411-413
68. Toubro, S., A. V. Astrup, L. Breum, and F. Quaade (1993). Safety and efficacy of long-term treatment with ephedrine, caffeine and an ephedrine/caffeine mixture. Int J Obes Relat Metab Disord; 17 Suppl 1:S69-72
69. Van Dyke, C., J. Ungerer, P. Jatlow, P. Barash, and R. Byck (1982). Intranasal cocaine: dose relationships of psychological effects and plasma levels. Int J Psychiatry Med; 12:1-13
70. Zeisel, S. H (1999). Regulation of "nutraceuticals". Science; 285:1853-1855
71. Zhang, X (1998). Regulatory situation of herbal medicines: A worldwide review. World Health Organization, p. 49
72. Spilker, B (1991). Guide to clinical trials. New York: Raven Press, 1156