
Division of Respiratory and Critical Care Physiology and Medicine, Harbor-UCLA Medical Center, 1000 W. Carson St., Torrance, CA 90509, USA.
Resumen
RELACIONES ENTRE EL ESTADO RED-OX EN EL CITOPLASMA Y EN LA MITOCONDRIA CON LA RESPIRACIÓN MITOCONDRIAL
El concepto desarrollado aquí es, que la misma cadena de transporte de electrones y la citocromooxidasa son responsables de la regulación del estado red-ox tanto del citoplasma como de la mitocondria. Mientras tanto, el estado red-ox de ambos compartimentos están determinados por la tasa relativa de reoxidación de la coenzimas oxidativas, tales como la nicotinamida adenina dinucléotido (NAD), solamente la NAD reducida (NADH) de la mitocondria tiene acceso directo a la cadena de transportes de electrones.
En contraste con esto, la reoxidación del NADH del citoplasma involucra la transferencia de su protón por el camino de un transportador de protones («H+ shuttle») a través de la membrana mitocondrial para reducir el NAD de la mitocondria, reoxidación que puede, luego, tomar lugar a través de la cadena común de transporte de electrones. Por lo tanto, los pasos de la reacción de reoxidación del NADH del citoplasma son mayores en número comparados con los del NADH mitocondrial.
Además, hay un efecto de capacitancia para el transporte de los protones producidos en el citoplasma porque estos iones deben ser transferidos de un relativamente grande compartimiento NAD/diluido en el citoplasma a un más pequeño compartimiento NAD/concentrado en la mitocondria. De esta forma, una constante de mayor tiempo (1/resistencia x capacitancia) para la reoxidación del NADH en el citoplasma, en relación a la mitocondria, podría causar un estado red-ox más bajo en el citoplasma en comparación con el compartimiento mitocondrial durante condiciones de «no estado estable» (non steady-state conditions).
La consecuencia de un estado red-ox disminuido del citoplasma deviene en una acumulación de ácido láctico, el cual, por mecanismos a describir en detalle más adelante, aumentará la PO2 capilar.
IMPORTANCIA DEL BICARBONATO (HCO3) COMO NEUTRALIZADOR O «BUFFER» DEL ÁCIDO LÁCTICO
La Figura 1 muestra cómo la glucólisis anaeróbica podría sostener la respiración mitocondrial y la producción de energía a tasas de trabajo intenso. Cuando aumenta el ácido láctico en la célula muscular, CO2 extra es producido por la célula como resultado de la acción buffer del HCO3– sobre el ácido láctico (Figura 1, paso 3). Debido a que el HCO3– disminuye y el lactato aumenta en la célula, los gradientes de concentración desarrollados entre el HCO3– y el lactato de sangre y célula inducen intercambios de estos aniones (Figura 1, paso 4). Este mecanismo satura y rellena el sutil sistema buffer de la célula.
Tanto el aumento de la producción celular no aeróbica de CO2, como la disminución en sangre del HCO3– , sirven para incrementar la concentración del ion de hidrógeno ([H+]) en el capilar, modificando hacia la derecha la curva de disociación de la oxihemoglobina. Este mecanismo aumenta la PO2 capilar y el potencial para difusión de O2, anticipando la anaerobiosis total y permitiendo una más completa extracción de O2 de la sangre, que de otra manera no es posible (Fig. 1, paso 5). De este modo, en el Síndrome de Mc Ardle, cuadro patológico en el cual la acidosis láctica no se desarrolla (7), la liberación de O2 por parte de la hemoglobina no está facilitada, y la diferencia arteriovenosa de O2, a niveles de máximo esfuerzo es patológicamente pequeña (alrededor de un 1/3 de lo normal)(6).
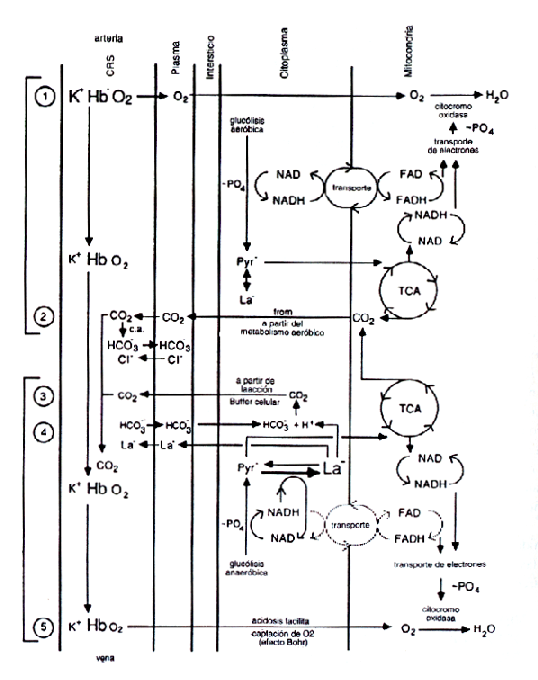
Figura 1. Pasos del intercambio de O2 y CO2 entre
células musculares y capilar, y producción de fosfágeno por parte del
metabolismo aeróbico, anaeróbico y del metabolismo aeróbico facilitado por el
desvío hacia la derecha de la curva de disociación de la oxihemoglobina (Hb O2)
inducida por la producción de H+ del ácido láctico (efecto Bohr) durante
el
ejercicio intenso. Mientras este esquema muestra cambios de la arteria a la
vena, estos podrían ser también esquematizados como diferencias entre las
relaciones cociente de alto flujo sanguíneo/consumo de O2 y bajo
flujo sanguíneo/consumo de O2 por unidad muscular. En la llave más
altas a la izquierda (incluye pasos 1 y2): los cambios en niveles de O2
y CO2 sanguíneos, secundarios al metabolismo aeróbico. En la llave
más baja a la izquierda incluye pasos 3, 4 y 5): cambios adicionales en los
niveles sanguíneos de O2 y CO2, secundarios al aumento de
producción de ácido láctico. PASO 1: Descarga del O2 de la
hemoglobina resultante de la disminución de la PO2 mitocondrial a
consecuencia de la fosforilación oxidativa de ADP. PASO 2: Reacciones que
involucran CO2 generadas por el metabolismo aeróbico. PASO 3: El CO2
generado por el HCO3– (Bicarbonato) celular a partir de la acción buffer
por la acumulación de ácido láctico (1 meq HCO3 =22.4 ml CO2).
El
CO2, desarrollado a partir de la neutralización de H+ con HCO3–
causa un gran incremento de la PCO2 y un descenso del pH sanguíneo. Esto
esta en contraste con el CO2 producido por el metabolismo aeróbico
para el cual, el concomitante descenso de la saturación de O2 de la
Hb minimiza el ascenso en la PCO2 (efecto Christiansen-Douglas-Haldane)
y el descenso del pH. PASO 4: El consumo de HCO3–
sanguíneo por el ácido láctico producido por las células genera un intercambio
equilibrado de lactato y HCO3– entre el plasma y la célula. PASO 5:
la PO2
aumenta, aunque la saturación de O2 disminuya (desvío de la curva de
disociación de la oxihemoglobina hacia la derecha), Esto ocurre por una
disminución del pH en los glóbulos rojos, consecuente a una disminución del HCO3–, un aumento del CO2, por la acción buffer de HCO3– sobre el
ácido láctico en las células musculares y la no presencia del efecto Haldane
disponible, para minimizar los cambios de pH por el CO2, generado por
la acción buffer, en contraste con aquéllos del metabolismo aeróbico.
Siglas: FAD=flavin-adenina-dinucleótido; FADH= FAD reducido (adiciona un
H+); CRS = células rojas de la sangre (eritrocitos); La-= ácido láctico; Pyr- =
piruvato; TCA= ciclo de los ácidos tricarboxílicos; NAD= nicotina-adenina-dinucleotido;
NADH= NAD reducido (adiciona un H+).
Los factores físico-químicos determinados por la constante disociación de ácido láctico (pK= 3.08) y el pH del citoplasma (~7,0) requieren que >99% del ácido láctico sea neutralizado (por buffer) inmediatamente después de su formación en la célula. La presión osmótica celular aumentará 17 mm Hg por aumento milimolar de lactato, a menos que un osmol, simultáneamente, desaparezca de la célula, a medida que el ácido láctico es neutralizado en ella. Esto está de hecho probado, porque el ácido láctico es neutralizado casi en su totalidad por la sustancia buffer volátil, el HCO3– . El incremento en la cantidad de enzimas en los músculos, hallada algunas veces ante ejercicios muy intensos, puede estar causada por el aumento de la presión osmótica celular, resultante de la neutralización de ácido láctico recientemente formado, a través de sustancias buffer no-inestables, cuando ya se depletó el HCO3– celular.
¿CUÁNTO EFECTO TIENE EL AUMENTO DEL ÁCIDO LÁCTICO SOBRE LA PO2 CAPILAR?
Nosotros podemos tomar sangre humana normal y ponerla en presencia de gases que contienen dos niveles de PO2, 35 y 15 mm Hg, y variados valores de PCO2, con el objeto de cambiar su pH, dentro de un rango que pudiese considerarse fisiológico. Luego, dividimos cada muestra tonometrada (es decir, cuantificada la presión parcial de gases), adicionando solución salina anaeróbicamente (3% por volumen) a una muestra, y un volumen igual de una solución de ácido láctico a otra muestra, y posteriormente, medimos las diferencias de PO2, pH y PCO2 entre las muestras comparadas. Este estudio mostró que la PO2 se incrementa en forma lineal con el descenso del pH, a dos niveles de PO2 (Figura 2A) en una cantidad que fue cercanamente predicha por la propuesta de Severinghaus (11).
Mientras que el incremento de PO2 fue menor para el decremento del pH, cuando la PO2 inicial fue baja (Figura 2A), el % de incremento de la PO2 por meq/l de lactato fue similar (Figura 2B). Dado que el incremento de lactato durante la parte más inclinada de la curva cinética de lactato puede ser de 6 meq/L/min o mayor, dependiendo de la tasa de trabajo, el aumento de la PO2 causado por el aumento de concentración de ácido láctico, es relativamente grande comparado con la PO2 capilar de los tejidos que están produciendo dicho ácido láctico.
Esto podría facilitar enormemente la difusión de O2 bajo condiciones de alta demanda del mismo. El VO2 con el cual este efecto comienza a hacerse evidente podría variar entre sujetos, dependiendo de su «umbral» láctacido (anaeróbico) o aptitud física para esfuerzos de endurance o resistencia. Además el ácido láctico producido por hipoxia, sirve para adaptarse a diferentes grados de hipoxia en los tejidos, haciendo posible rendir altos niveles de trabajo, aunque con tiempos de rendimiento en resistencia reducidos.
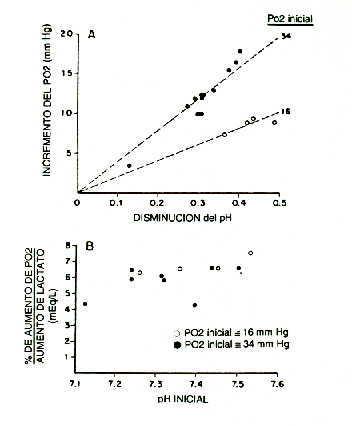
Figura 2. Medición del efecto Bohr a PO2 de 34 y 16 mm Hg,
respectivamente. Incrementos en la PO2 causados por la adición
anaeróbica de solución de ácido láctico (vs. solución salina normal) en muestras
comparables de sangre humana fresca tonometrada.
Parte A= aumento de PO2 (y) inducido por la declinación del pH (x).
Para una PO2 inicial de 34 mmHg en 12 muestras, y= 39,6.x, r= 0,655.
Además, ante un incremento del pH de 0,30 unidades, el efecto Bohr aproxima en
12 y 6 mmHg la PO2 inicial de 34 y 16 mm Hg, respectivamente. Parte
B= el % de incremento de la PO2, por cada 1 meq/1 de incremento de
lactato en relación al pH inicial. A estos valores simulados de la PO2
capilar, el efecto Bohr es aproximadamente de un 6% por cada meq/l de incremento
de lactato, indistinto en relación a cualquier pH inicial.
¿POR QUÉ EL O2 TRANSPORTADO A LAS CÉLULAS EN RESPUESTA AL EJERCICIO NECESITARÍA UN PROCESO DE FACILITACIÓN?
Esta pregunta estaría dirigida más específicamente a intensidades de trabajo elevado. A partir de la ecuación para difusión [VO2 = k (Pc-Pm) A/L, donde: A es área de difusión, L es distancia de difusión, Pc es PO2 capilar, Pm es la PO2 mitocondrial y K es una constante que refleja la difusibilidad del O2], es evidente que el incremento de la masa de O2 transferida que se necesita para regenerar ATP a tasas de trabajo progresivamente mayores requiere que el cociente entre numerador y denominador en la ecuación de difusión aumente en forma lineal con el consumo de O2 (VO2).
El aumento de la conductancia vascular es el mayor mecanismo en el incremento agudo del área de superficie de difusión y el decremento de la distancia de difusión. Sin embargo, como la conductancia en el sistema vascular no se incrementa en forma lineal con el aumento de la tasa de esfuerzo, sino lentamente por encima de tasas moderadas de trabajo (3), el mantenimiento, y eventualmente, el aumento en la diferencia de PO2 entre el capilar y la mitocondria deberá ocurrir en conducir más O2 hacia el interior de la mitocondria ante el requerimiento de una tasa incrementada de O2, necesaria para ejecutar un ejercicio intenso.
Mientras el suministro de O2 (QO2= flujo sanguíneo x contenido arterial de O2) se incrementa, normalmente, en forma lineal con el VO2 (consumo de O2) (Figura 3), el cociente entre QO2 y consumo muscular de O2 (VO2 m) disminuye.
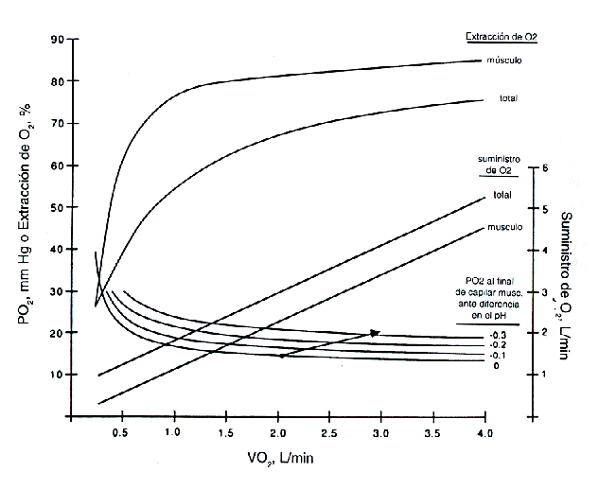
Figura 3. Relación entre el transporte de O2 total (volumen
minuto x contenido arterial de oxígeno) y el transporte de O2
muscular (flujo sanguíneo muscular x contenido arterial de oxígeno), durante el
ejercicio en función del consumo de O2, extracción de O2
muscular y total, y PO2 en el final del capilar muscular. Véase el
texto para conocer los datos utilizados para calcular las curvas. La flecha
sobre las curvas de PO2 en el final del capilar, ilustran el aumento
en PO2 (de 13 a 19 mmHg) como resultado del incremento de lactato
sanguíneo en 6 meq/1, en la transición durante la cual el 1% O2
aumenta de 2 L/min a 3 L/min.
El decremento en la relación QO2/VO2m determina un incremento de la extracción de O2 (Figura 3).
Dado que el contenido capilar de O2 en el final del tramo venoso desciende con el incremento del trabajo muscular, aun con la necesidad de regenerar ATP más rápidamente, el incremento del flujo en masa de O2 hacia la mitocondria por proceso de difusión, puede ocurrir solamente si la PO2 del capilar muscular es mantenida o incrementada.
A fin de ilustrar cuantitativamente el balance de los requerimientos de provisión de O2 y el cambio de la PO2 durante el ejercicio, para un varón normal de tamaño promedio, fue construida la Figura 3, asumiendo que el volumen minuto cardíaco se incrementa linealmente con el VO2, con una pendiente de ≈6 (9), a una concentración de hemoglobina de 15 g/dl y a una saturación de O2 (SaO2) de 95 %.
Las presunciones son a partir de una tasa metabólica de reposo total del cuerpo de 0.250 l/min, con un volumen minuto cardíaco de 5 L/min, de los cuales 20 % contribuye a la irrigación de músculos esqueléticos, donde todo el incremento de VO2 y volumen minuto cardíaco durante el ejercicio, representa un incremento de circulación sanguínea y del metabolismo de los músculos en ejercicio. También, se asume, que la tasa neta metabólica y la circulación sanguínea de los restantes órganos del cuerpo permanecen sin cambios durante el ejercicio.
Si el sujeto realiza un ejercicio a un VO2 = 2 L/m (por ej., VO2 m = 1,8 L/min), y no hay cambios en la SaO2 (saturación de O2 en sangre arterial) o en la concentración de hemoglobina, el flujo de O2 muscular podría ser de 2,2 L/min. Por lo tanto, la sangre al final del capilar en los músculos ejercitados sería saturada con O2, solamente en un 18 %, y simultáneamente la saturación de O2 en sangre venosa mixta sería del 32 % (Figura 3). Si este trabajo fuese hecho sin acidosis láctica, la PO2 al final del capilar en el músculo sería solamente 13-14 mm Hg (Figura 3, mirar final de la flecha) (10). Sin embargo, el sujeto realizó un ejercicio con una tasa de esfuerzo con un costo en VO2 de 3 L/min, durante el cual el lactato aumenta 6 meq/l, como se ilustra en la Fig. 4 (parte superior), describiéndose la dinámica del lactato durante el mismo, la PO2 al final del capilar se incrementaría en un ≈6 mmHg (Figura 3, flecha), o el 40 % (basados en el cambio de PO2, resultante de la adición anaeróbica de ácido láctico a la sangre venosa; (Figura 2).
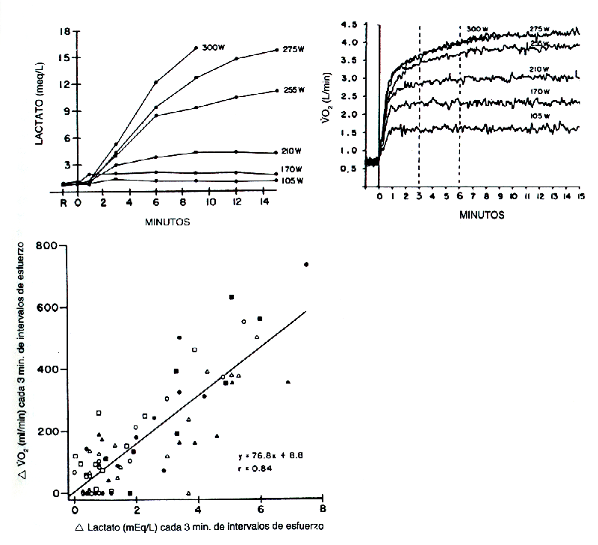
Figura 4. Arriba: mediciones simultáneas de VO2 y cinética del lactato en
vena del brazo, relacionadas con el tiempo a varias velocidades de trabajo de
bicicleta ergométrica realizado durante 15 min o hasta extenuación (datos de la
Ref. 8). Abajo: relación entre el incremento de lactato cada 3 min de intervalo
de esfuerzo, y el incremento de VO2 en el mismo período de tiempo, en
trabajos en cicloergómetro, realizados durante 15 minutos o hasta la extenuación.
Los resultados pertenecen al sujeto ilustrado en el gráfico de arriba y de otros
5 sujetos normales, mientras realizaron algunos protocolos de ejercicio (datos
de la Ref. 8).
Además, a pesar del bajo contenido de O2, la PO2 al final del capilar en los ejercicio musculares, puede ser más alta aun VO2 de 3 L/min que a un VO2 de 2 L/ min, como se observa en la Figura 3, creando una fuerza potencial para incrementar el flujo en masa de O, hacia el interior de los músculos.
Dado que una mitocondria aislada puede producir ATP a presiones parciales tan bajas como 1 mm Hg, algunos investigadores consideran que, en tanto que algo de O2 pueda ser medido en los tejidos, la disponibilidad de O2 raramente, limita la regeneración de ATP. Por supuesto, estas PO2 estimadas son valores medios para un número de células y muchas mitocondrias (2), no tomándose en cuenta la heterogeneidad metabólica en tejidos de metabolización rápida, como fue descrito por Chance (1). Si el O2 no puede alcanzar la mitocondria lo suficientemente rápido, la PO2 en algunas mitocondrias podría ser críticamente baja, a pesar de una PO2 tisular media >1 mm Hg.
Para transportar O2 desde los capilares a la mitocondria debe haber siempre un gradiente de presión entre la fuente de O2 (el capilar) y el recipiente de O2 (la mitocondria). Después de una vasodilatación máxima durante un ejercicio intenso, a mayor necesidad de O2, mayor debe ser el gradiente de PO2 entre capilares y mitocondria. Dado que altas presiones se requieren para mover una mayor masa de O2 por unidad de tiempo, el concepto de una simple PO2 capilar crítica es insostenible.
¿POR QUÉ CONSIDERAR QUE EL ÁCIDO LÁCTICO PUEDE TENER UN ROL ESPECIAL DURANTE EL EJERCICIO?
La remarcablemente suave pendiente de 5 L/min (6), 6 L/min (9), para el incremento del volumen minuto cardíaco, en función del VO2 (L/min), en respuesta al ejercicio, indica que el índice QO2/VO2 musculares bastante uniforme.
Asimismo, el relativamente constante umbral de ácido láctico en un individuo dado, al margen de la tasa a la cual el esfuerzo es incrementado (14), sugiere una remarcable uniformidad en la relación QO2/VO2 y que, en un individuo dado, la mayoría de las células musculares en ejercicio desarrollan acidosis al mismo nivel de consumo de O2 (VO2). Sin embargo, cuando la relación muscular QO2/VO2 regional se torna demasiado baja para obtener los requerimientos energéticos celulares, la generación regional de ácido láctico incrementa la PO2 local por el mecanismo mostrado en la Figura 1, y modula el efecto de la QO2, relativamente baja, incrementando la extracción local de O2.
¿DINÁMICA DEL CONSUMO DE OXÍGENO Y PRODUCCIÓN DE LACTATO: «CORRELACIÓN INOCENTE» O «RELACIÓN CAUSA Y EFECTO»?
Como describimos previamente, el VO2 (consumo de O2) y la dinámica del lactato aparentan estar interrelacionadas durante el esfuerzo intenso. La Figura 4, gráfica comparativamente, varias concentraciones simultáneas de lactato tomadas de la vena antecubital, ante diferentes cargas de trabajo constante de 15 minutos de duración o a nivel de fatiga, para un solo sujeto.
Para el más bajo nivel de trabajo, el consumo (VO2) permanece estable por 3 minutos y no hay un sostenido crecimiento de la concentración de lactato. En tanto la tasa de trabajo se incrementa, el estado de equilibrio del VO2 se demora y el lactato aumenta.
El lactato parece alcanzar un valor constante solamente cuando el VO2 alcanza un valor constante. Esto está mostrado en la Figura 4 (abajo), donde se relacionan los cambios en concentración de lactato y el aumento del VO2, cada 3 minutos de intervalo, durante ejercicios de 15 minutos de duración, con los que se evaluo al sujeto; esto se ilustra en la parte alta de la Figura 4; en la parte baja hay estudios similares en otros 5 sujetos.
Luego de cada período de 3 minutos, hay una buena correlación entre el aumento de VO2 y el de lactato, sin importar la duración del ejercicio ni la intensidad del trabajo. Nuestra hipótesis es que, después de 3 minutos de ejercicio, el incremento en el VO2 se hace posible por la producción de ácido láctico en las células hipóxicas. Esto sirve para desviar la curva de disociación de la oxihemoglobina a la derecha, aumentando la PO2 de los capilares, permitiendo a su vez, el aumento de VO2 en función de los requerimientos musculares.
Respecto a esto, el aumento de ácido láctico es responsable por el lento incremento de VO2 que se observa con tasas de trabajo por encima del umbral de acidosis láctica, y la correlación observada al pie de la Figura 4 es un mecanismo causa-efecto, y no una relación espuria. Además la acidosis láctica producida por anaerobiosis sirve para disminuir el estado anaeróbico.
MECANISMO POR EL CUAL LA FORMACIÓN A DISTANCIA DE PIRUVATO NEUTRALIZA EL ESTADO RED-OX EN EL CITOPLASMA EN MÚSCULOS BAJO UN PROCESO DE GLUCÓLISIS ANAERÓBICA
La glucólisis puede ser aeróbica sin un cambio en el estado red-ox del citoplasma, cuando el transporte de protones de la membrana mitocondrial se mantiene en estado estable, por las coenzimas mitocondriales y la cadena de transporte de electrones (Figura 1, lado arterial o alto Q/VO2 m). Cuando el transporte de H+ no es adecuadamente reoxidado por los mecanismos oxidativos de la mitocondria, la glucólisis anaeróbica causa incremento del ácido láctico (Figura 1, lado venoso o bajo Q/VO2 m). Cuando aumenta el lactato, se difunde en la sangre donde es transportado a células con lactato relativamente bajo y un alto estado red-ox. Cuando el lactato entra a dichas células, será convertido a piruvato, apropiado al estado red-ox de esa célula. Por lo tanto, el piruvato de la sangre aumenta, pero tardíamente y a una tasa más lenta que el incremento de lactato (13).
Cuando este piruvato, recientemente generado, circula hacia los músculos en ejercicio, entra en células con bajo piruvato, reduciendo de este modo el cociente lactato/piruvato y NADH/NAD en el citoplasma. Este piruvato puede servir como sustrato hidrocarbonado para el ciclo de Krebs de las células musculares metabólicamente activas, sin consumir NAD adicional del citoplasma.
EL SÍNDROME DE Mc ARDLE COMO MODELO DE UN DEFECTO EN EL MECANISMO DE FACILITACIÓN DE LA EXTRACCIÓN DE O2 POR LA ACIDOSIS LÁCTICA
Los pacientes con síndrome de Mc Ardle se caracterizan por sufrir limitaciones en el ejercicio debidas a dolores musculares, a menudo acompañados por subsiguiente mioglobinuria, reportadas como una deficiencia en la fosforilasa muscular (6). Las limitaciones al ejercicio ocurren usualmente con un VO2 de ~1 L/min o al ~40-50 % del VO2 máx., previamente predicho en los pacientes. El único rasgo distintivo de este síndrome es que el ácido láctico no aumenta durante el ejercicio, ni siquiera ante la capacidad máxima de trabajo del sujeto. Sin embargo la inosina y la hipoxantina sí aumentan, de la misma forma como se comportan en sujetos normales en su capacidad máxima de trabajo (12), sugiriendo una regeneración de ATP disminuida.
La fisiopatología del síndrome de McArdle va más allá del defecto de la fosforilasa muscular. Estos pacientes tampoco pueden extraer O2 eficientemente durante el ejercicio (6). Además, la diferencia arteriovenosa de O2 en su máxima capacidad de trabajo, es marcadamente reducida comparada con sujetos normales, alcanzando un valor máximo de solamente alrededor de 6 ml/dl en tanto, en sujetos normales rondan los 15 ml/dl (6). Nosotros postulamos que, en el síndrome de Mc Ardle, el mecanismo y la falla para extraer O2 está ligado a la falla de producción de ácido láctico. Esto limita las posibilidades para que el paciente mantenga un adecuado PO2 capilar para desempeñar con normalidad ejercicios intensos. Consistente con la hipótesis de que los pacientes con síndrome de Mc Ardle no pueden extraer una cantidad normal de O2 de la sangre durante el ejercicio, por su falla para aumentar el ácido láctico en las células musculares, presentamos evidencias como: 1) que desarrollan una marcada taquicardia de esfuerzo, alcanzando frecuencias cardíacas máximas, aun durante ejercicios por debajo del VO2 máx. predicho y medido; 2) que el volumen minuto cardíaco aumenta en relación a su VO2, aproximadamente 2,5 veces de lo habitualmente normal (6). El mecanismo para este inusualmente alto volumen minuto cardíaco observado en estos pacientes sería compensatorio y similar a aquéllos hallados en otras condiciones, en los cuales la diferencia arteriovenosa de O2 es reducida (ej., hipoxia celular muscular relativa), tal los casos de una anemia, carboxihemoglobinemia e hipoxemia arterial.
Mientras la depleción de glucógeno en sujetos normales no previene el incremento de lactato, a igual nivel de VO2 durante ejercicios, en los cuales se incrementa el lactato en estados de «no deplección de glucógeno», el incremento absoluto de lactato a tasas de trabajo máximo esta reducido. Una disminución del VO2 máx. acompaña está reducción en el lactato máximo. Esto coincidiría con el rol del ácido láctico como facilitador de la obtención de O2 por las células que lo necesitan.
En síntesis, parecería que el desarrollo de la acidosis láctica fuera esencial para la performance a tasas de esfuerzo por sobre el umbral de acidosis láctica. El H+ producido localmente con el lactato, en condiciones de flujo de O2 inadecuado a los tejidos, mejora la PO2 capilar y facilita la difusión de O2 a la mitocondria. Este mecanismo sirve para proveer un centro de retroalimentación en condiciones de alta necesidad de O2 e hipoxia tisular, cuando existe un disbalance entre los requerimientos y la provisión de VO2.
La correlación entre VO2 y la dinámica del lactato ante ejercicios de elevada intensidad es consistente con el concepto de que la producción de ácido láctico facilita el consumo muscular de O2. Por lo tanto, el lento crecimiento de VO2 que se observó a tasas de trabajo constantes por sobre el umbral de acidosis láctica puede ser explicado por el incremento de la PO2 creado por el desvío de la curva de disociación de la oxihemoglobina hacia la derecha (efecto Bohr), como resultado del incremento neto local del ácido láctico.
Es como un componente esencial del ejercicio normal por sobre el umbral de acidosis láctica, sin lo cual esfuerzos a esos niveles no serían posible. El síndrome de McArdle nos provee un modelo clínico ejemplificador del estado del metabolismo muscular y la limitación de la tolerancia al ejercicio que ocurre cuando está ausente el mecanismo facilitador del ácido láctico.
REFERENCIAS
1. Chance, B (1989). Metabolic heterogeneities in rapidly metabolizing tissues. J. Appl. Cardiol. 4: 207-221
2. Connett, R.J., T.E.J. Gayeski, and C.R. Honig (1986). Lactate efflux is unrelated to intracellular PO2, in a working red muscle in situ. J. Appl. Physiol. 61: 402-407
3. Higginbotham, M.B., K.G. Morris, R.S. Williams, P.A. McHale, R.E. Coleman, and F.R. Cobb (1986). Regulation of stroke volume during submaximal and maximal upright exercise in normal man. Cir. Res. 58: 281-291
4. Hill, A. V., C.N. Long, and H.Lupton (1924). Muscular exercise, lactic acid, and the supply and utilization of oxygen. VI. The oxygen debt at the end of exercise. Proc. R. Soc. Lond. 97: 127-137
5. Jobsis, F.F., and W.N. Stainsby (1968). Oxidation of NADH during contractions of circulated mammalian skeletal muscle. Respir. Physiol. 4: 292-300
6. Lewis, S.F., and R.G. Haller (1986). The pathophysiology of McArdles disease: clues to regulation in exercise and fatigue. J. Appl. Physiol. 61: 391-401
7. Ross, B.D., G.K. Radda, D.G. Gadian, G. Rocker, M. Esiri, and J. Falconer-Smith (1981). Examination of a case of suspected McArdles syndrome by 31 P nuclear magnetic resonance. N. Engl. J. Med. 304: 1338-1342
8. Roston, W.L., B.J. Whipp, J.A. Davis, D.A. Cunningham, R.M. Effros, and . Wasserman (1987). Oxygen uptake kinetics and lactate concentration during exercise inhuman. Am Rev. Respir. Dis. 135: 1080-1084
9. Rowell, L.B (1986). Human Circulation Regulation During Physical Stress. New York: Oxford Univ. Press, p. 215
10. Severinghaus, J.W (1979). Simple accurate equation for human blood O2 dissociation computation. J. Appl. Physiol. 46: 599-602
11. Severinghaus, J.W (1976). Acid-base balance nomogram – a Boston-Copenhagen detente. Anesthesiology 45: 539-541
12. Sinkeler, S., E. Joosten, R. Wevers, R. Binkhorst, and L. Oei (1986). Skeletal muscle adenosine, inosine, and hypoxanthine release following ischemic forearm exercise in myoadenylate deaminase deficiency and McArdles disease. Adv. Exp. Med. Biol. B. 195: 517-523
13. Wasserman, K., W.L. Beaver, J.A. Davis, J.Z. Pu, D. Heber, and B.J. Whipp (1985). Lactate, pyruvate, and lactate-to-pyruvate ratio during exercise and recovery. J. Appl. Physiol. 59: 935-940
14. Wasserman, K., W.L. Beaver, and B.J. Whipp (1990). Gas exchange theory and the lactic acidosis (anaerobic) threshold. Circulation 81, Suppl. 11:11-14-11-30
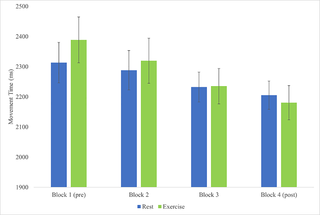
Un episodio agudo de ejercicio aeróbico reduce el tiempo de movimiento en una tarea de Fitts
El movimiento tiene una importancia crucial en el control motor, reflejado a través del tiempo de...
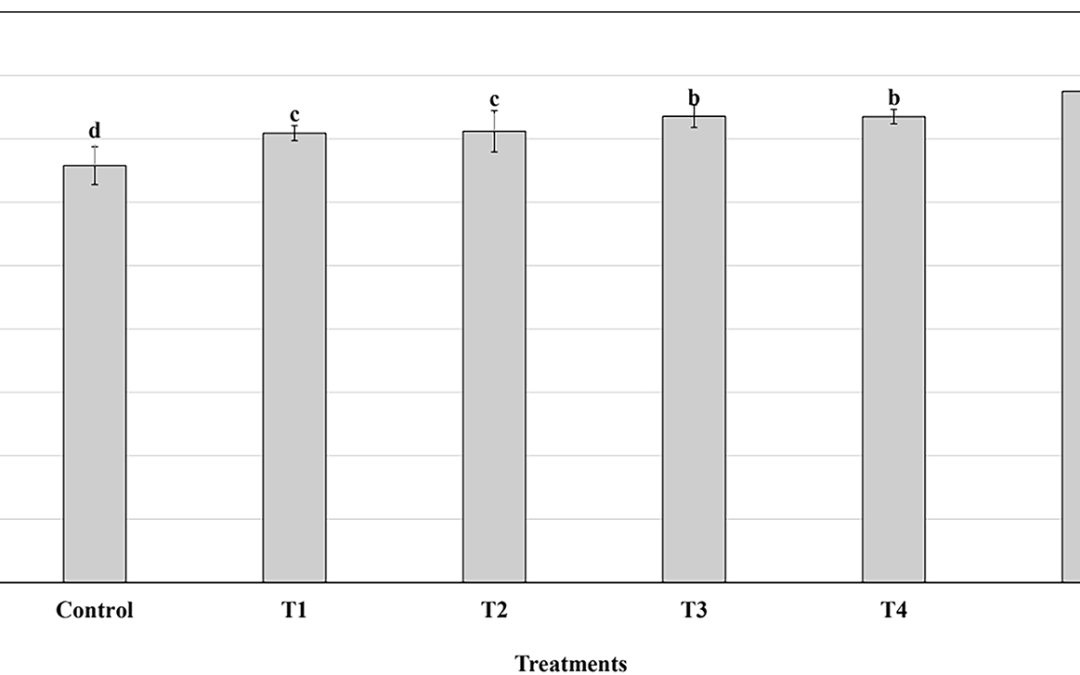
Mejora de las propiedades nutricionales y de textura del jerky reestructurado con polvo de sangre de pato para aplicaciones en kits de comida
Resumen del Estudio sobre el Uso de Polvo de Sangre de Pato en Jerky Reestructurado En un contexto...
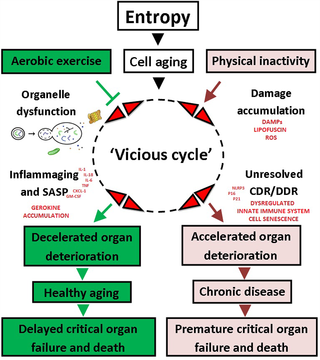
El ejercicio aeróbico a lo largo de la vida protege contra la inflamaging y el cáncer
Ejercicio Aeróbico y su Impacto en el Envejecimiento y el Riesgo de Cáncer La investigación...
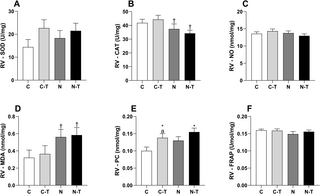
La combinación de nandrolona y entrenamiento de resistencia indujo remodelación cardíaca y estrés oxidativo a pesar de la contractilidad mejorada de los cardiomiocitos
Este artículo investiga cómo la combinación de entrenamiento de resistencia (RT) y el uso de...
El efecto del ciclismo en la función cognitiva y el bienestar en adultos mayores
El impacto del ciclismo al aire libre en la función cognitiva y bienestar de los adultos mayores...