
Fatiga Muscular: ¿La Causa Principal es el Ácido Láctico o los Fosfatos Inorgánicos?
Håkan Westerblad1, David G Allen2 y Jan Lännergren1
1Department of Physiology and Pharmacology, Karolinska Institutet, SE-171 77 Stockholm, Sweden.
2Department of Physiology and Institute of Biomedical Research, University of Sydney F13, New South Wales 2006, Australia.
Artículo publicado en el journal PubliCE, Volumen 0 del año 2002.
Publicado 15 de septiembre de 2003
Resumen
Palabras clave: lactacidemia, fosfato inorgánico, acidosis
INTRODUCCIÓN
El consumo de energía de las células del músculo esquelético puede incrementarse más de 100 veces durante la transición del descanso al ejercicio de alta intensidad. Esta alta demanda de energía excede la capacidad aeróbica de las células musculares, y una gran parte del ATP requerido provendrá del metabolismo anaeróbico. El ejercicio de alta intensidad también lleva a una rápida disminución de la función contráctil conocida como fatiga del músculo esquelético. Por lo tanto parece lógico que existe una relación causal entre el metabolismo anaeróbico y la fatiga muscular; es decir, alguna(s) consecuencia(s) del metabolismo anaeróbico causa la disminución de la función contráctil.
La ruptura anaeróbica del glucógeno lleva a una acumulación intracelular de ácidos inorgánicos, de los cuales el ácido láctico es cuantitativamente el más importante. Debido a que el ácido láctico es un ácido fuerte, éste se disocia en lactato e H+. Los iones de lactato tendrían un pobre efecto sobre la contracción muscular (16); sin embargo, el incremento de los H+ (i.e., la reducción del pH o producción de acidosis) es la causa clásica de fatiga en el músculo esquelético. Sin embargo el rol de una reducción del pH como una causa importante de fatiga está siendo actualmente puesto en duda, y varios estudios recientes (5, 14, 19, 20) muestran que una reducción del pH puede tener un escaso efecto sobre la contracción en músculos de mamíferos a temperaturas fisiológicas.
Además de la acidosis, el metabolismo anaeróbico en el músculo esquelético también incluye la hidrólisis de fosfato de creatina (CrP) a creatina y fosfato inorgánico (Pi). La creatina tiene un escaso efecto sobre la función contráctil, mientras que existen varios mecanismos a través de los que el incremento del Pi puede reducir la función contráctil. Así, sobre la base de hallazgos recientes (6, 8, 10, 12), el incremento del Pi más que la acidosis parece ser la causa más importante de fatiga durante en el ejercicio de alta intensidad. Esta breve revisión resumirá los resultados que conforman las bases del cambio que sustentan al incremento en el Pi y no a la acidosis como la causa principal de fatiga en el músculo de los mamíferos. Nos concentraremos en los estudios en los que la fatiga se desarrolló sobre la escala de tiempo de minutos, ya que en este tiempo las consecuencias del metabolismo anaeróbico pueden ser de importancia superlativa. Con una activación constante más intensa (e.g., una contracción máxima continua), otros factores, como la falla en la propagación del potencial de acción, se pueden volver de importancia creciente. Contrariamente, en ejercicios de mayor duración (e.g., carrera de maratón), causas como la depleción en los depósitos de carbohidratos y la deshidratación se vuelven de importancia creciente.
Para estudiar los mecanismos subyacentes de la fatiga, nosotros frecuentemente usamos células musculares aisladas (fibras), las cuales son fatigadas por tétanos repetidos de breve duración. La presente revisión se concentrará sobre los resultados obtenidos en ese tipo de trabajos como también en estudios realizados sobre fibras musculares sin sarcolema (i.e., células musculares dónde la membrana celular ha sido química o físicamente removida). Esto es debido a que los estudios sobre fibras musculares individuales proveen el camino más directo para identificar los mecanismos celulares de la fatiga. Puede ser argumentado que las conclusiones derivadas de estudios realizados en fibras individuales no son relevantes a la fatiga experimentada por humanos durante diversos tipos de ejercicio. Sin embargo, los datos disponibles indican que los mecanismos de la fatiga son cuantitativamente similares en diferentes modelos experimentales, desde el ejercicio en humanos, hasta una fibra individual (2). Las diferencias que inevitablemente deben existir parecen ser principalmente de una naturaleza cuantitativa.
La elevación y caída del ácido láctico como una causa directa de la disfunción del músculo esquelético en la fatiga
Durante la actividad muscular intensa el pH intracelular puede caer en ~0.5 unidades de pH. Existen dos importantes líneas de evidencia que han sido utilizadas para relacionar esta caída en el pH con la disfunción contráctil en la fatiga. Primero, los estudios sobre fatiga muscular en humanos han mostrado frecuentemente una buena correlación temporal entre la caída del pH muscular y la reducción en la producción de fuerza o potencia. Segundo, los estudios en fibras del músculo esquelético sin sarcolema han demostrado que la acidificación puede reducir tanto la fuerza isométrica como la velocidad de acortamiento muscular.
Sin embargo, en humanos la correlación temporal entre el deterioro de la función contráctil durante la fatiga y el pH reducido no está siempre presente. Por ejemplo, la fuerza generalmente se recupera más rápidamente que el pH después de finalizar contracciones musculares que inducen fatiga (18). Esto significa que si un pH reducido tiene un efecto directo en la disminución de la fuerza en músculos de humanos, este efecto debe estar contrarrestado por algún otro factor que incremente la fuerza en igual medida. Debido a que no se ha identificado un factor potenciador de la fuerza, la conclusión obvia es que no existe una relación causal entre la acidosis y la reducción en la producción de fuerza.
La evidencia importante a favor de la acidosis como causante de la reducción de la producción de fuerza proviene de estudios en fibras musculares sin sarcolema realizados en temperaturas < 15 °C (14). Estudios recientes se han concentrado en la incidencia que ejerce la temperatura sobre los efectos del pH en la fuerza, y los resultados de estas investigaciones fomentaron la objeción sobre el rol del H+ en la fatiga muscular de mamíferos. Algunos estudios realizados hace más de 10 años mostraron que la acidificación, si es que la hubo, resultó en un incremento en la fuerza tetánica a temperaturas fisiológicas (17). Más recientemente, Pate y colegas (14) estudiaron las fibras del psoas sin sarcolema de un conejo y observaron el gran efecto depresivo de un pH disminuido a 10 °C, pero el efecto de la acidificación sobre la producción de fuerza fue menor a 30°C. Subsecuentemente se han obtenido resultados similares en las fibras musculares aisladas de rata y en el músculo entero de una rata (29) (Fig 1 A).
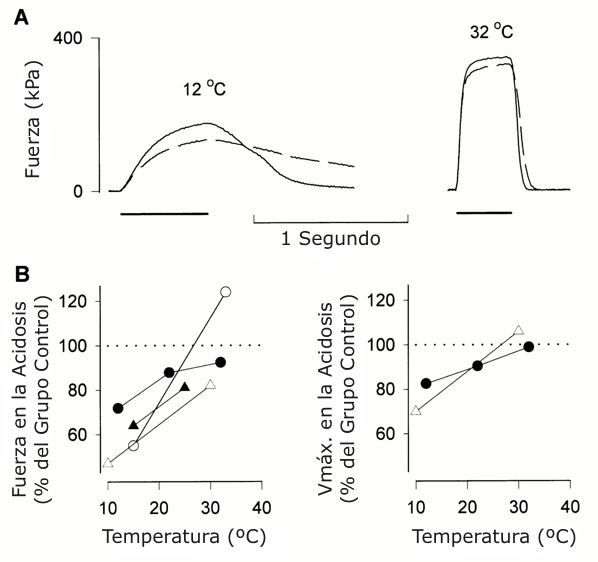
Figura 1. A: registros de las fuerzas originales de la contracción
tetánica producida en una fibra muscular intacta del músculo de la pata de una
rata. Los registros fueron obtenidos bajo condiciones controladas (línea sólida)
y cuándo la fibra fue acidificada por ~0.5 unidades de pH a través del
incremento del baño de CO2 (línea discontinua). Notar que los efectos de la
acidificación son marcadamente mayores a 12°C que a los 32°C. El período de
estimulación está indicado debajo de los registros de fuerza. B: los efectos
depresivos de la acidosis sobre la producción de fuerza (izquierda) y sobre la
máxima velocidad de acortamiento muscular (Vmáx, derecha) disminuyen cuándo la
temperatura es incrementada. Los datos fueron obtenidos de fibras intactas de un
ratón (Ref. 19), fibras sin sarcolema de un conejo (Ref. 14), y del músculo
intacto de una rata (Ref. 20). Las líneas punteadas indican que no hay
diferencias entre acidosis y control. A esta adaptada de la Ref. 19.
La acidificación ha sido considerada como un importante factor detrás de la reducción de la velocidad de acortamiento muscular durante la fatiga. Sin embargo, utilizando fibras musculares sin sarcolema de un conejo, Pate y colegas (14) demostraron que la acidificación tiene un escaso efecto sobre la velocidad de acortamiento muscular a 30°C. Similarmente en fibras musculares intactas de un ratón, la máxima velocidad de acortamiento fue reducida en un 20% a 12°C mientras que no hubo reducciones significativas a 32°C (19). Por ello en el músculo de mamíferos estudiados a temperaturas fisiológicas, la función de los puentes cruzados (i.e., el acoplamiento y desacoplamiento cíclico de las cabezas de miosina con la actina que se producen durante la contracción muscular) está poco afectada por la acidificación (Fig 1B).
Otro mecanismo por el cual la acidosis intracelular puede inducir fatiga esta dado por la inhibición del metabolismo energético. Las enzimas claves en la glucogenolisis y la glucólisis son la fosforilasa y la fosfofructo quinasa, respectivamente. Estas enzimas son inhibidas a un pH bajo in vitro, y por ello la tasa de aprovisionamiento de ATP a los procesos demandantes de energía [e.g., el ciclo de los puentes cruzados y el bombeo de Ca2+ del retículo sarcoplasmático] podrían estar disminuidos en músculos que se vuelven ácidos durante la fatiga. Sin embargo, un estudio reciente en humanos falló en detectar una reducción en la tasa glucogenolítica/glucolítica en músculos acidificados (4). Además, una acidificación de 0.4 unidades pH no afectó la resistencia de fibras musculares aisladas de un ratón fatigadas por tétanos breves repetidos a 28°C (5). Así, la inhibición de la fosforilasa y la fosfofructo quinasa inducida por acidosis in vitro, aparentemente está contrarrestada in vivo por otros factores, y el desarrollo de la fatiga no parece estar acelerado por la acidosis a temperaturas cercanas a las fisiológicas. También se ha sugerido que la acidosis disminuye el rendimiento muscular durante la fatiga a través de la inhibión en la liberación de Ca2+ desde el retículo sarcoplasmático. Tal inhibición puede disminuir el grado de activación de la maquinaria contráctil y así generar una caída en la producción de fuerza. Aunque se ha mostrado repetidamente que hay una caída en la concentración de Ca2+ mioplasmático libre ([Ca2+]i) durante las contracciones en presencia de fatiga (ver Figura 2A), es poco probable que esto esté relacionado a la acidosis. Un hallazgo que puede avalar el rol de la caída del pH en este aspecto es que la acidosis reduce la probabilidad de apertura de los canales del retículo sarcoplasmático aislado, para la liberación de Ca2+ (i.e. los receptores de rianodina). Sin embargo, la acidificación no tiene un efecto reductor obvio sobre la despolarización inducida por la liberación de Ca2+ del retículo sarcoplamsático, en fibras sin sarcolema con un sistema de túbulos transversos del retículo sarcoplasmático intacto (13).
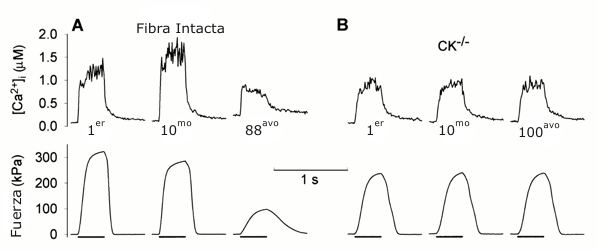
Figura 2. Fuerza típica y concentración de Ca2+ mioplasmático libre
([Ca2+]i) datos obtenidos durante la fatiga inducida por tétanos breves
repetidos en una fibra normal (A) y en una fibra con déficit completo de creatín
quinasa (CK-/-, B). La fibra normal mostró los cambios característicos, con un
incremento temprano de la [Ca2+]i acompañado por una pequeña reducción en la
fuerza. Posteriormente siguió una caída en la [Ca2+]i tetánica y la fuerza hasta
que la estimulación es detenida después de 88 tétanos. Contrariamente, ni la
[Ca2+]i tetánica ni la fuerza es alterada durante 100 tétanos en la fibra CK-/-,
la que se fatigó sin la acumulación de fosfato inorgánico (Pi). Notar también la
menor fuerza en el estado no fatigado en la fibra CK-/-, la cual tenía una mayor
concentración de Pi en reposo. Los períodos de estimulación están indicados
debajo de los registros de fuerza. Figura adaptada de la Ref. 7.
Para resumir la acidosis tiene un escaso efecto directo sobre la producción de fuerza isométrica, la máxima velocidad de acortamiento muscular, o la tasa de ruptura de glucógeno en los músculos de mamíferos estudiados a temperaturas fisiológicas. Por ello, si la acidosis está envuelta en la fatiga del músculo esquelético, el efecto debería ser indirecto. Por ejemplo, la acidosis extracelular puede activar en un buen grado el grupo III y IV de nervios aferentes en el músculo y por ello estar incluida en la sensación de disconfort durante la fatiga. Esto puede ser bien percibido desde el punto de vista de un atleta. Los regímenes de entrenamiento para los atletas de alto nivel en deportes de resistencia generalmente enfatizan “el entrenamiento de ácido láctico”, (i.e., protocolos de entrenamiento que inducen una alta concentración de niveles de lactato plasmático). Un efecto de este tipo de entrenamiento puede ser aprender a enfrentarse con el disconfort inducido por la acidosis sin perder el ritmo y la técnica y de esta manera conseguir el máximo efecto fuera de los músculos, los cuales en sí mismos no están directamente inhibidos por la acidosis. Un mecanismo alternativo a través del cual la formación de ácido láctico puede imponer un límite sobre el rendimiento es durante los ejercicios de larga duración en los cuales la depleción de glucógeno es clave. Con una producción extensiva de ácido láctico, la cantidad total de ATP producido desde los depósitos de glucógeno es menor que con la ruptura aeróbica completa, debido a que cada unidad de glucosa entrega 3 ATP cuándo el ácido láctico es producido y 39 ATP cuando es completamente metabolizado en la mitocondria a CO2 y H2O. Por ello los depósitos de glucógeno son depletados más rápidamente cuándo se producen grandes cantidades de ácido láctico mientras que el rendimiento muscular disminuye severamente a bajas concentraciones de glucógeno. Finalmente, la correlación temporal frecuentemente observada entre la caída del pH y la disminución de la función muscular puede ser más coincidente que causal. Una marcada acidificación implica que la demanda de energía excede la capacidad del metabolismo aeróbico y que las vías anaeróbicas son usadas para generar ATP. Puede ser, entonces, que más que la acidificación, alguna otra con consecuencia del metabolismo anaeróbico sea el verdadero factor del deterioro de la función muscular, y el incremento del Pi es un fuerte candidato en lo que respecta a este tema.
El incremento de la acumulación de Pi como la causa principal de fatiga en el músculo esquelético
La concentración de Pi se incrementa durante la actividad intensa del músculo esquelético principalmente debido a la ruptura de CrP. Muchos modelos de acción de los puentes cruzados proponen que se libera Pi en la transición de los estados de baja fuerza y acoplamiento débil a los estados de alta fuerza y acoplamiento fuerte. Esto implica que la transición al estado de alta fuerza esta dificultada por un aumento en el Pi. Por ello, menos puentes cruzados podrían permanecer en estado de alta fuerza y la producción de fuerza podría caer con el incremento Pi durante el desarrollo de la fatiga. En línea con estas investigaciones en fibras sin sarcolema se mostró consistentemente una reducción en la activación máxima forzada de Ca2+ en presencia de una elevada concentración de Pi.
La hipótesis acerca de que el incremento del Pi reduce la fuerza máxima de los puentes cruzados ha sido difícil de evaluar en células musculares intactas, ya que dificultosamente se ha probado incrementar el Pi mioplasmático sin producir a la vez otros cambios metabólicos. Nosotros demostramos recientemente (6,7) que ratas genéticamente modificadas en ausencia completa de creatín quinasa (CK) en sus músculos esqueléticos (CK-/- rata) proveen un modelo razonable para estudiar los efectos del incremento del Pi proveniente de la transferencia del grupo fosfato de alta energía entre la PCr y el ATP catalizado por la CK. Durante períodos de alta demanda de energía, el resultado de la reacción de la CK es la ruptura del CrP a Cr y Pi, pero la concentración de ATP permanece casi constante. Las fibras rápidas del músculo esquelético CK-/- de una rata muestran un incremento en la concentración Pi mioplasmático en reposo; además, durante la fatiga no hay una acumulación significativa de Pi. La activación máxima forzada de Ca2+ de las fibras rápidas CK-/- no fatigadas es marcadamente menor que las fibras normales, las que soportan el rol del incremento del Pi, el cual disminuye la fuerza (6). Además, durante la fatiga inducida por tetanos breves repetidos, las fibras rápidas con la CK intacta mostraron una reducción de 10-20% de la máxima fuerza de activación rápida de Ca+2, después de ~10 tétanos. Esta disminución de la fuerza, la que ha sido atribuida al incremento del Pi, no ocurre en las fibras CK-/- (7). Del mismo modo después de 100 tétanos, la fuerza no fue significativamente afectada en las fibras CK-/-, mientras que en este momento estuvo reducida <30% de la original en las fibras normales (Figura 2). Un sustento adicional para la conexión entre la concentración Pi mioplasmático y la producción de fuerza en células musculares intactas proviene de investigaciones en las cuales la concentración mioplasmática disminuida de Pi está asociada con el incremento en la producción de fuerza (15). Así, el incremento en el Pi mioplasmático puede disminuir la producción de fuerza durante la fatiga por una acción directa sobre la función de los puentes cruzados. La función alterada de los puentes cruzados puede también afectar la relación de fuerza–[Ca2+]i a través de la compleja interacción entre el acoplamiento de los puentes cruzados y la activación de los filamentos de actina. En este camino, el incremento del Pi puede también reducir la producción de fuerza causando una reducción de la sensibilidad miofibrilar al Ca2+, lo cual es una característica observada en el músculo esquelético fatigado.
En años recientes, se ha vuelto más claro que el incremento en el Pi también afecta el desarrollo de la fatiga debido a la acción sobre el manejo del Ca2+ del retículo sarcoplasmático. Con respecto a esto, hay muchos mecanismos a través de los cuales el aumento del Pi puede forzar estos efectos, y los resultados pueden tanto, incrementar o reducir la [Ca2+]i tetánica. Los mecanismos importantes son los siguientes:
Acción Directa
El Pi puede actuar directamente sobre los canales de liberación de Ca+2 del retículo sarcoplamático, incrementar su probabilidad de apertura y facilitar la liberación inducida de Ca+2 (3). Esta acción del Pi podría llevar a un incremento de la [Ca2+]i tetánica y podría estar incluido en el incremento de la [Ca2+]i tetánica normalmente observado en la fatiga temprana. En respaldo a esta idea, las fibras CK-/- no muestran este incremento temprano en la [Ca2+]i tetánica.
Inhibición del consumo de Ca2+
El aumento del Pi puede inhibir el ATP accionado para el consumo de Ca2+ del retículo sarcoplasmático (SR) (9). En el corto término, la inhibición del consumo de Ca2+ del SR puede resultar en un incremento de la [Ca2+]i tetánica (asumiendo que la cantidad de liberación de Ca2+ permanece constante). En el largo plazo, por otro lado, el Ca2+ podría acumularse en otras organelas (e.g. mitocondria) o posiblemente salir de la célula. En este caso el Ca2+ disponible para la liberación podría caer sustancialmente, resultando en una reducción de la [Ca2+]i tetánica. Aunque es teóricamente posible que la pérdida de Ca2+ de la célula contribuya a la caída de la [Ca2+]i tetánica en la fatiga, no estamos enterados de ningún hallazgo experimental que sostenga esta teoría.
Precipitación Ca2+-Pi
El Pi puede entrar en el SR, lo que puede resultar en la precipitación C2+-Pi y por ello disminuir el C2+ disponible para la liberación. Este mecanismo ha ganado apoyo recientemente de estudios que usaron varias aproximaciones experimentales diferentes. En los experimentos iniciales en fibras sin sarcolema con el sistema de túbulos transversos del SR intacto, Fryer y colegas (10) demostraron que el incremento del Pi puede disminuir la liberación de Ca2+ del SR. Estos autores también proveyeron evidencia directa acerca de que el Pi puede alcanzar la concentración suficientemente alta en el SR para exceder el umbral para la precipitación de Ca2+-Pi en este entorno, con elevada concentración de Ca2+. Desde este trabajo pionero, se ha demostrado que la disponibilidad de Ca2+ para la liberación está en realidad reducida en fibras individuales fatigadas de los músculos de un sapo (11). Las mediciones de la concentración de Ca2+ del SR también mostraron una caída en las fibras fatigadas del sapo (12). Además, la caída de la [Ca2+]i tetánica durante la fatiga esta retardada cuándo la acumulación de Pi está prevenida por la inhibición de la reacción de la CK, ya sea farmacológicamente (8) o por la supresión genética (CK-/-) (7).
Una debilidad de la hipótesis de que el incremento del Pi causa una precipitación Ca2+-Pi en el SR es que el Pi se incrementa más bien al inicio durante la estimulación pero la caída de la [Ca2+]i tetánica generalmente ocurre un poco más tarde. Por otra parte, en las fibras rápidas del ratón la caída en la [Ca2+]i tetánica correlacionan temporalmente con un incremento en el Mg2+, lo cual presumiblemente se origine de una ruptura neta de ATP (2), y la relación entre la precipitación Ca2+-Pi en el SR y el incremento de Mg2+/reducción de ATP no es obvia. Sin embargo un estudio reciente provee una razonable explicación para estas aparentes dificultades: el Pi probablemente entre en el SR vía un canal de aniones, el cual incrementa su apertura probablemente a medida que el ATP disminuye (1). Esto puede explicar como el Pi entra al SR con un retraso y por que existe una correlación temporal entre el incremento del Mg2+ y la caída en la [Ca2+]i tetánica. Interesantemente, en las fibras en las que la reacción de la CK esta farmacologicamente inhibida y la fatiga ocurre sin una gran acumulación de Pi, un incremento en Mg2+ no está acompañado por una reducción de la [Ca2+]i tetánica (8). Todos los resultados obtenidos con una variedad de aproximaciones experimentales indican que la precipitación Ca2+-Pi en el SR es la causa principal de la reducción de la [Ca2+]i tetánica en la fatiga inducida por tetanos breves repetidos.
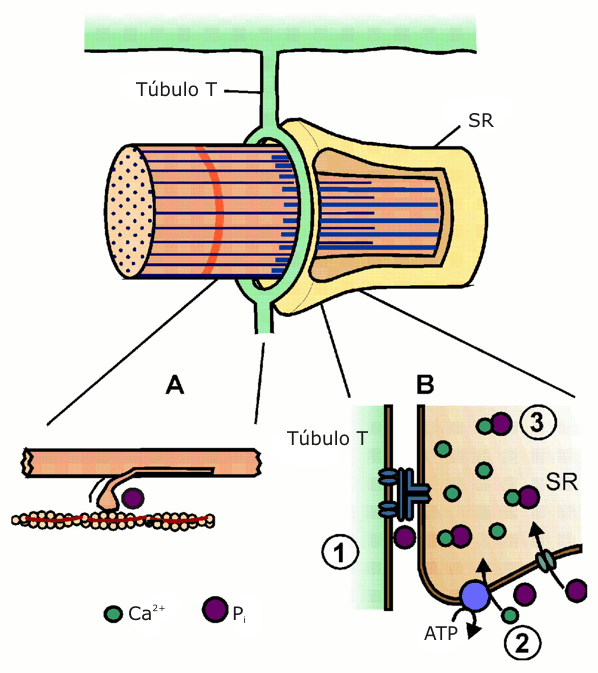
Figura 3. Ilustración de los sitios dónde el incremento del Pi puede
afectar la función contráctil durante la fatiga. El incremento en el Pi puede
actuar directamente sobre las miofibrillas y disminuir la producción de fuerza
de los puentes cruzados y la sensibilidad miofibrilar al Ca2+ (A). Actuando
sobre el manejo de Ca2+ (B) del retículo sarcoplasmático (SR), el aumento del Pi
puede también incrementar la [Ca2+]i tetánica en la fatiga temprana, a través de
la estimulación de los canales de liberación de Ca+2 del SR (1); inhibiendo la
captación de ATP-Ca2+ conducido al SR (2); y reduciendo la [Ca2+]i tetánica en
la fatiga tardía entrando en el SR, precipitando con Ca2+, y por ello
disminuyendo Ca2+ disponible para la liberación (3).
La Figura 3 ilustra varios mecanismos a través de los cuales el Pi puede afectar la función muscular durante la fatiga. La Figura muestra que el incremento del Pi puede disminuir la producción de fuerza actuando directamente sobre las miofibrillas o sobre los sitios de excitación-contracción dentro de las células musculares. El efecto depresivo del Pi puede, igual que el efecto de la acidificación descripto anteriormente, disminuir cuándo la temperatura esta incrementada a nivel del que prevalece en músculos de mamíferos in situ. Existe poca información disponible acerca de cómo los efectos del incremento del Pi sobre la contracción muscular son influenciados por la temperatura, y muchos estudios en fibras sin sarcolema, que analizaron los efectos del Pi, se han realizado a temperaturas bajas, los estudios realizados en fibras intactas de ratón en nuestro laboratorio mostraron un marcado efecto depresivo sobre la producción de fuerza que puede ser atribuido a la elevación del Pi. Estos estudios se han realizado generalmente a ~25°C, lo que esta cerca de la temperatura in situ en reposo (31°C) de los músculos superficiales situados en el pie, utilizados en el estudio (5). Sin embargo se necesitan realizar estudios sobre músculos de mamíferos a una temperatura corporal normal (~37°C) para estar seguros que los efectos del Pi permanecen cuando la temperatura es incrementada.
Conclusión
Los datos presentados anteriormente proveen un sustento substancial del rol clave del incremento del Pi en la fatiga del músculo esquelético. Para la acidosis, por otro lado, los datos más recientes indican que su efecto depresivo sobre la contracción muscular es limitado.
Agradecimientos: Agradecemos a Britta Flock por construir la Figura 3. Nuestra investigación fue financiada por el Centro Nacional de Investigación Deportiva, y el Consejo Nacional de Salud e Investigaciones Médicas de Australia.
Referencias
1. Ahern GP and Laver DR (1998). ATP inhibition and rectification of a Ca2+-activated anion channel in sarcoplasmic reticulum of skeletal muscle. Biophys J 74: 23352351
2. Balog EM, Fruen BR, Kane PK, and Louis CF (2000). Mechanisms of Pi regulation of the skeletal muscle SR Ca2+ release channel. Am J Physiol Cell Physiol 278: C601C611
3. Bangsbo J, Madsen K, Kiens B, and Richter EA (1996). Effect of muscle acidity on muscle metabolism and fatigue during intense exercise in man. J Physiol (Lond) 495: 587596
4. Dahlstedt AJ, Katz A, and Westerblad H (2001). Role of myoplasmic phosphate in contractile function of skeletal muscle: studies on creatine kinase-deficient mice. J Physiol (Lond) 533: 379388
5. Dahlstedt AJ, Katz A, Wieringa B, and Westerblad H (2000). Is creatine kinase responsible for fatigue? Studies of skeletal muscle deficient of creatine kinase. FASEB J 14: 982990
6. Dahlstedt AJ and Westerblad H (2001). Inhibition of creatine kinase reduces the rate of fatigue-induced decrease in tetanic [Ca2+]i in mouse skeletal muscle. J Physiol (Lond) 533: 639649
7. Duke AM and Steele DS (2000). Characteristics of phosphate-induced Ca 2+ efflux from the SR in mechanically skinned rat skeletal muscle fibers. Am J Physiol Cell Physiol 278: C126C135
8. Fryer MW, Owen VJ, Lamb GD, and Stephenson DG (1995). Effects of creatine phosphate and Pi on Ca2+ movements and tension development in rat skinned skeletal muscle fibres. J Physiol (Lond) 482: 123140
9. Kabbara AA and Allen DG (1999). The role of calcium stores in fatigue of isolated single muscle fibres from the cane toad. J Physiol (Lond) 519: 169176
10. Kabbara AA and Allen DG (2001). The use of fluo-5N to measure sarcoplasmic reticulum calcium in single muscle fibres of the cane toad. J Physiol (Lond) 534: 8797
11. Lamb GD, Recupero E, and Stephenson DG (1992). Effect of myoplasmic pH on excitation-contraction coupling in skeletal muscle fibres of the toad. J Physiol (Lond) 448: 211224
12. Pate E, Bhimani M, Franks-Skiba K, and Cooke R (1995). Reduced effect of pH on skinned rabbit psoas muscle mechanics at high temperatures: implications for fatigue. J Physiol (Lond) 486: 689694
13. Phillips SK, Wiseman RW, Woledge RC, and Kushmerick MJ (1993). The effect of metabolic fuel on force production and resting inorganic phosphate levels in mouse skeletal muscle. J Physiol (Lond) 462: 135146
14. Posterino GS, Dutka TL, and Lamb GD (2001). L(+)-lactate does not affect twitch and tetanic responses in mechanically skinned mammalian muscle fibres. Pflügers Arch 442: 197203
15. Ranatunga KW (1987). Effects of acidosis on tension development in mammalian skeletal muscle. Muscle Nerve 10: 439445
16. Sahlin K and Ren JM (1989). Relationship of contraction capacity to metabolic changes during recovery from a fatiguing contraction. J Appl Physiol 67: 648654
17. Wiseman RW, Beck TW, and Chase PB (1996). Effect of intracellular pH on force development depends on temperature in intact skeletal muscle from mouse. Am J Physiol Cell Physiol 271: C878C886