
Metabolismo de las Grasas Durante el Ejercicio Una Revisión. Parte II: Regulación del Metabolismo y los Efectos del Entrenamiento
Asker Jeukendrup1, William H Saris1 y Anton J Wagenmakers1
Nutrition Research Center, Department of Human Biology, Maastricht University, Maastricht, The Neterlands.
Artículo publicado en el journal PubliCE, Volumen 0 del año 1999.
Publicado 16 de julio de 2007
Resumen
Palabras clave: entrenamiento, ciclo glucosa-ácido graso, malonil CoA, epinefrina, insulina, intensidad de ejercicio
INTRODUCCION
En la parte I de esta revisión, “Movilización de ácidos grasos y metabolismo muscular” publicada en una edición previa del Internacional Journal of Sports Medicine (70), se hizo una reseña de la importancia de las grasas como sustrato durante el ejercicio, y se describió que nivel de grasas se moviliza desde el tejido adiposo, cuanto de ese monto es transportado a través de la sangre, tomado por el músculo y utilizado en el mismo para su oxidación. Además se discutieron los roles de diferentes combustibles lípidos (ácidos grasos en plasma, ácidos grasos derivados de lipoproteínas plasmáticas, ácidos grasos de triacilglicéridos intramusculares). La parte II se concentrará en la interacción entre el metabolismo de carbohidratos y grasas, y la regulación de la utilización de sustratos. Además se discutirá el efecto de la intensidad del ejercicio y el entrenamiento. En la parte III: “Efecto de las intervenciones nutricionales”, en una edición posterior de esta publicación (71), se le prestará atención a los efectos de las diversas manipulaciones nutricionales sobre el metabolismo de los ácidos grasos.
REGULACION DE LA UTILIZACION DE SUSTRATOS
Regulación hormonal
Los cambios en la gluconeogénesis, lipólisis y cetogénesis durante el ejercicio pueden ser explicados, al menos en parte, por los cambios en las concentraciones de hormonas. El perfil catabólico hormonal, tal como se observa durante el ejercicio de resistencia intenso se promoverá luego de una dieta baja en carbohidratos o con un balance de energía negativo (40, 41, 77, 80). Las hormonas pueden afectar primariamente la movilización de ácidos grasos (e.g., lipólisis). La concentración de insulina decrece mientras que los niveles de glucagón, epinefrina, norepinefrina, así como también la hormona de crecimiento (STH) y cortisol, se incrementan. La STH tiene un efecto facilitador sobre las catecolaminas, y por lo tanto también sobre la liberación de ácidos grasos en el torrente sanguíneo. Las catecolaminas tienen un potente efecto estimulante sobre la lipólisis, mientras que la insulina es un fuerte inhibidor de la lipólisis (48) (ver también parte I de esta revisión: Sección “Lipólisis en el tejido adiposo”. Galbo (40) concluyó que los cambios en las concentraciones plasmáticas de estas hormonas fueron principalmente causadas por cambios en las concentraciones plasmáticas de glucosa. Sin embargo, no puede excluirse una estimulación simpática de liberación de hormonas. Durante el ejercicio las catecolaminas plasmáticas y la actividad simpática neural se elevan exponencialmente con el incremento de la intensidad de ejercicio. Sin embrago, el efecto de las concentraciones de catecolaminas en el plasma sobre la lipólisis y la oxidación de grasas durante el ejercicio no han sido muy bien descritas. Romijn et al (102) demostraron que durante el ejercicio a baja intensidad (25 % VO2 máx) la renovación (turnover) de ácidos grasos se incrementó cinco veces, mientras que las concentraciones de catecolaminas en plasma se elevaron solo 50 % por sobre los valores de reposo. Cuando se incrementó la intensidad del ejercicio hasta el 65 y 85 % del VO2 máx los niveles de catecolaminas en plasma se incrementaron 3-6 y 7-19 veces, respectivamente, pero la renovación de ácidos grasos en realidad decreció. Por lo tanto, otros factores como el flujo sanguíneo del tejido adiposo (11, 12), la concentración de lactato en plasma (67), la concentración plasmática de insulina (63) y el incremento del flujo glucolítico (111) pueden desempeñar un rol principal durante el ejercicio a intensidades moderadas a normales. Se revisó extensivamente en diversas publicaciones recientes el rol de las hormonas durante el ejercicio y su influencia en la utilización de sustratos (34, 38, 39, 100, 120).
El ciclo glucosa-ácidos grasos en reposo
La interacción entre el metabolismo de grasas y los carbohidratos puede ser explicado en parte por la existencia del llamado ciclo glucosa-ácidos grasos propuesto por Randle en 1963 (94) (Figura 1). Un incremento en la concentración de ácidos grasos en plasma podría conducir a una oxidación de grasas aumentada. El flujo acelerado a través de la vía de la beta-oxidación puede resultar en una acumulación de acetil CoA y NADH, lo cual en cambio podría inhibir la actividad de la enzima piruvato dehidrogenasa (PDH) y por lo tanto inhibir la oxidación de piruvato. La inhibición de la piruvato dehidrogenasa podría conducir a un ahorro de carbohidratos, dado que el piruvato convertido en acetil CoA, y es sometido a oxidación. Adicionalmente, una concentración incrementada de acetil CoA y de hecho un aumento en el cociente acetil CoA/CoA resultaría en un incremento de la concertación de citrato conduciendo a una inhibición de la enzima fosfofructokinasa (PFK), una enzima llave (determinante de la velocidad) en la vía glucolítica. Estos efectos podrían resultar en una reducción de la tasa de glucólisis y glucogenólisis. Concentraciones mas bajas de ácidos grasos pueden generar, por supuesto, las adaptaciones opuestas.
Si bien se obtuvo evidencia a favor de la existencia de este ciclo glucosa-ácidos grasos en el músculo cardíaco de ratas (42, 43, 94, 95) y en el diafragma de ratas (42, 43, 94, 95) estudios iniciales en músculos esqueléticos de ratas sugirieron que la disponibilidad de ácidos grasos no inhibe la disponibilidad o la oxidación de glucosa (8, 49, 101). Sin embargo, los resultados fueron más bien conflictivos, dado que otros estudios reportaron una disminución de la utilización de glucosa luego de adicionar ácidos grasos al medio de perfusión en el músculo esquelético de ratas aislado (91, 98, 99). Rennie et al (99) hallaron que el incremento en la disponibilidad de ácidos grasos disminuyó la tasa de degradación de glucógeno en músculos esqueléticos estimulados de ratas. Las concentraciones de citrato se incrementaron en los músculos de fibras lentas ST; sóleo) y rápidas semi-oxidativas (FTOx; porción profunda del vasto lateral), pero no en los músculos de fibras rápidas glucolíticas (FTGl; porción superficial del vasto lateral). Por eso la diferencia en los estudios mencionados previamente puede ser, en parte, explicada por los diferentes tipos de músculos estudiados. Otra diferencia entre los estudios, podría ser el momento de medición. Zorzano et al (130) concluyeron que el ciclo glucosa-ácidos grasos no opera en estado de reposo en el sóleo de una rata privada de alimentos, pero opera en el período post ejercicio.
Si bien la mayoría (5, 35, 36, 115, 123, 129), pero no todos, los estudios proveen datos que respaldan la existencia del ciclo de Randle en el músculo esquelético en reposo, este tema seguirá siendo un asunto de debate continuo. Ferrannini et al (36) demostraron que los niveles elevados de ácidos grasos en plasma inhibieron el consumo de glucosa, hallazgo que fue confirmado posteriormente por Walter et al (123). Estudios en los cuales se investigó la tolerancia a la glucosa con altos y bajos niveles de ácidos grasos plasmáticos, sugieren un efecto inhibidor directo de los ácidos grasos sobre el transporte de glucosa (35). Wolfe et al (128), por otro lado, observaron que los niveles elevados de ácidos grasos no afectaron la oxidación de glucosa en plasma cuando el consumo de esta se mantuvo constante. En cambio, los niveles elevados de ácidos grasos suprimieron la oxidación de glucógeno, Baron et al (7) administraron “Intralipid” y heparina, y observaron que la utilización corporal total de glucosa mediada por insulina se inhibió en reposo.
La mayoría de los estudios en sujetos humanos en reposo sostienen el concepto de que un incremento en la disponibilidad de ácidos grasos afecta la utilización de glucosa intracelular o extracelular, o ambas. Sin embargo, los mecanismos mencionados son, en gran medida, desconocidos, y si bien hay indicadores de que el ciclo de Randle es operativo en el músculo esquelético de humanos en reposo, hay poco respaldo para el ciclo en condiciones de ejercicio.
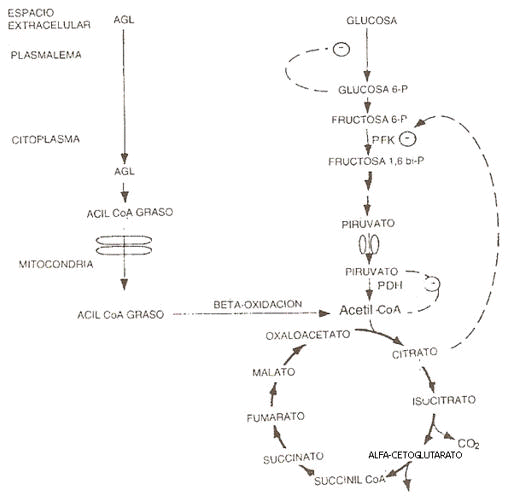
Figura 1. Ciclo glucosa-ácidos grasos (Ciclo de Randle). Este ciclo
describe los mecanismos por los cuales la oxidación de ácidos grasos puede
inhibir la oxidación de glucosa. Cuando los ácidos grasos entran al citoplasma
estos pueden ser activados a acil CoA graso, y subsecuentemente transportados
dentro de la mitocondria. Vía beta-oxidación las unidades de acil CoA graso se
transformarán en acetil CoA. Una concentración aumentada de acetil CoA
citoplasmático o del cociente acetil CoA/CoA inhibirá la actividad de la PDH, y
por lo tanto inhibirá la decarboxilación del piruvato a acetil CoA. Además una
concentración incrementada de citrato puede conducir a la inhibición de la PFK.
Concentraciones aumentadas de glucosa-6-P citoplasmática pueden inhibir el
transporte de glucosa dentro de la célula muscular. (PDH = piruvato
dehidrogenasa; PFK = fosfofructokinasa; P = fosfato).
El ciclo glucosa-ácidos grasos durante el ejercicio
En uno de los primeros estudios que analizaron el ciclo glucosa-ácidos grasos en humanos durante la realización de ejercicio, se observó que el consumo de glucosa durante el ejercicio fue inhibido por un incremento en las concentraciones plasmáticas de ácidos grasos (23). En un clásico estudio, Costill et al (23) alimentaron a sus sujetos con una comida alta en grasas y les dieron una infusión de heparina para elevar los ácidos grasos en plasma. Los sujetos se ejercitaron 30 min sobre una cinta ergométrica al 70 % VO2 máx. La elevación de los ácidos grasos plasmáticos de aproximadamente 1 mmol/L redujo la tasa de ruptura de glucógeno muscular en un 40 %, comparado con la prueba control en la cual la concentración de ácidos grasos en plasma fue de aproximadamente 0.2 mmol/L. Vulkovich et al (121) observaron efectos similares de ahorro de glucógeno con alimentación con grasas y la infusión de “Intralipid”, en combinación con infusión de heparina. Estos hallazgos fueron además confirmados por Dyck et al (30) quienes infundieron “Intralipid” y heparina para elevar los ácidos grasos en plasma, observando un ahorro significativo de glucógeno luego de 15 min de pedaleo al 85% VO2 máx. Sin embargo, este último estudio no observó ninguna diferencia en el citrato muscular, acetil CoA y PDH entre las pruebas con “Intralipid” y las de control, sugiriendo que otro mecanismo que no es el ciclo glucosa-ácidos grasos debe ser el responsable de la regulación de la utilización de combustibles. Los autores sugieren una regulación al nivel de la glucógeno-fosforilasa (30). Hargreaves et al (51) elevaron las concentraciones de ácidos grasos en plasma por infusión de “Intralipid” y heparina. Durante el ejercicio (extensión de rodilla), se midieron la diferencia arterio-venosa de diferentes sustratos y se tomaron biopsias musculares. Ellos observaron que el consumo de glucosa fue inhibido, mientras que no pudieron ser encontradas diferencias ni en el índice de intercambio respiratorio del músculo ni en los niveles de glucosa-6-fosfato. Las concentraciones elevadas de ácidos grasos no resultaron en una disminución de la ruptura de glucógeno. Los autores sugirieron que los ácidos grasos tienen un efecto directo sobre el consumo de glucosa del espacio vascular. Estos resultados se obtuvieron durante ejercicios de baja intensidad, sin cambios en las concentraciones hormonales (51). Sin embargo, Romijin et al (103) demostraron que durante el ejercicio intenso, el consumo de glucosa no fue inhibido por las concentraciones aumentadas de ácidos grasos en plasma. Asimismo, para demostrar la contradicción de los hallazgos, Ravussin et al (96) no pudieron encontrar un cambio en la contribución relativa de grasas y carbohidratos a la oxidación total, durante el ejercicio al 44 % VO2máx, aunque los ácidos grasos plasmáticos estuvieron significativamente elevados por el suministro de una comida pre-ejercicio que contenía triacilglicéridos de cadena media (TCM).
Los resultados de diversos estudios están lejos de ser consistentes, especialmente durante el ejercicio (17, 23, 55, 96, 98, 99). Sin embargo, alguna de las diferencias en los resultados puede ser explicada por el diseño experimental. Algunos estudios fueron realizados en situaciones con tasas muy bajas de oxidación de ácidos grasos (79). Estas bajas tasas de oxidación de ácidos grasos se pueden encontrar, por ejemplo, durante muy bajas o muy altas intensidades de ejercicio. Algunos estudios administraron solo pequeñas cantidades de grasa exógena, lo cual puede haber sido insuficiente para elevar significativamente la oxidación de grasas (19, 79). Otros factores también pueden haber causado la no uniformidad en los resultados. Adicionalmente, el ciclo glucosa-ácidos grasos, probablemente, esté sujeto a la influencia de diversas hormonas, lo que hace dificultosa la comparación de diferentes fuentes de investigación. En la literatura, los diferentes hallazgos de estudios, algunos de los cuales observaron “ahorro de glucógeno” y otros que no, son generalmente explicados por el grado al cual se elevaron los niveles de ácidos grasos plasmáticos.
Sin embargo, puede ser más importante el grado al cual los niveles de ácidos grasos plasmáticos descendieron en la situación de control. Si el músculo es privado de ácidos grasos plasmáticos, como combustible (concentración baja, aproximadamente 0.2 mmol/L), el nivel de energía de la célula muscular puede estar reducido, conduciendo a una glucogenólisis acelerada.
Otra vía para observar los resultados posiblemente conflictivos es que el efecto de ahorro de glucógeno observado en algunos de los estudios no es el resultado de una reducción en la ruptura de glucógeno, sino mas bien una aceleración de la ruptura de glucógeno en las pruebas control. Estudios en los cuales se observó ahorro de glucógeno con niveles elevados de ácidos grasos (23, 30, 121), tuvieron niveles muy bajos de ácidos grasos plasmáticos en sus grupos control (por debajo de 0.2 mmol/L), lo cual puede haber privado al músculo de un sustrato plasmático. Por otro lado, los niveles de AGL en los estudios que no fueron capaces de demostrar este efecto, fueron de alguna manera mas altos (por encima de 0.4 mmol/L) (96). Esto es análogo al hallazgo de que el ácido nicotínico, un fuerte inhibidor de la movilización de ácidos grasos, que hace decrecer la disponibilidad de ácidos grasos usualmente por debajo de 0.2 mmol/L, incrementó la glucogenólisis muscular (9, 92). Esta visión es sostenida, además, por observaciones de Dyck et al (30) quienes demostraron un pobre nivel de energía en el músculo (bajo AMP y PCr) en los grupos control en los cuales la disponibilidad de ácidos grasos fue baja, comparado con las pruebas de “Intralipid” con concentraciones muy altas de ácidos grasos plasmáticos.
En conclusión, no hay evidencia de que el ciclo glucosa-ácidos grasos, tal como se propuso originalmente, sea operativo en el músculo esquelético de humanos en ejercicio. Si bien diversos estudios demostraron una disminución en la utilización de glucosa cuando los ácidos grasos son elevados, la regulación puede no ser a través del clásico ciclo glucosa-ácidos grasos. Por lo tanto la pregunta que subsiste es: si el ciclo glucosa-ácidos grasos no es el mecanismo regulador durante el ejercicio, ¿qué es lo que determina entonces la utilización de grasas y carbohidratos?
Regulación a través de la Malonil CoA
Otro posible mecanismo es la regulación a través de la Malonil CoA. Se sugirió que la carnitin-acil-transferasa I (CAT I), el paso tasa-limitante en el transporte de AGCL-CoA (AGCL = Ácidos Grasos de Cadena Larga) dentro de la mitocondria, es un importante sitio de regulación de la oxidación de ácidos grasos (Figura 2). La actividad de la CAT I, en cambio, es regulada por la Malonil CoA, la cual se forma por la acetil CoA-carboxilasa (ACC) en el paso tasa-limitante de la síntesis de ácidos grasos. La CAT I es muy sensible a cambios en las concentraciones de Malonil CoA (83). En ratas privadas de alimentos y diabéticas, los niveles de Malonil CoA hepático están reducidos y la oxidación de ácidos grasos, así como también la cetosis, están incrementadas (85). En situaciones de escasez de glucosa como el ayuno, la ACC es inactivada, y el resultado es una disminución en la concentración de Malonil CoA y un incremento de la tasa de beta-oxidación. Luego de la realimentación, las ratas previamente privadas de alimentos, incrementaron los niveles de Malonil CoA y redujeron la oxidación de ácidos grasos (85).
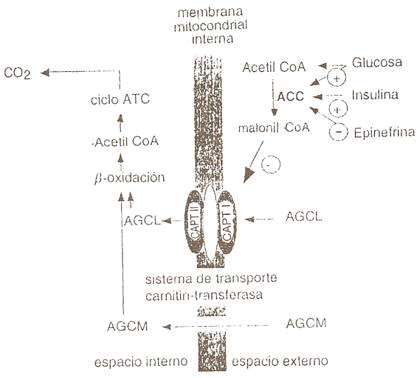
Figura 2. Mecanismos por los cuales la Malonil CoA puede regular el flujo de
ácidos grasos dentro de la mitocondria. La glucosa y la insulina pueden
estimular la acetil CoA-carboxilasa (ACC) conduciendo a una acumulación de
Malonil CoA, a cambio de lo cual se puede inhibir el transporte de ácidos grasos
de cadena larga (AGCL), carnitina-dependientes, a través de la membrana
mitocondrial. El transporte de ácidos grasos de cadena media (AGCM) a través de
la membrana mitocondrial, la que es menos sensible a los cambios en la
concentración de Malonil CoA, puede ser inhibido en un menor grado (25).
Estas observaciones sugieren que la Malonil CoA puede desempeñar un rol importante en la regulación de la oxidación de ácidos grasos (84). Si bien estos efectos se demostraron en estudios en el hígado (82, 84) y en el corazón (105), también existe evidencia de la regulación a través de la Malonil CoA en el músculo esquelético (31, 84, 106-108, 124, 135). La evidencia recogida en el músculo esquelético aislado y prefundido sugiere que la disponibilidad de carbohidratos puede ser un factor importante que determina la utilización de ácidos grasos (107). En hígado, corazón y también en músculo, los incrementos en las concentraciones de glucosa e insulina, y especialmente las dos en combinación, conducirán a una actividad incrementada de ACC y a un aumento en la formación de Malonil CoA (28, 107). Además, se ha demostrado que el acetoacetato (108), el ejercicio (124, 125) y las condiciones inducidas por estimulación eléctrica (28, 107) reducen en forma aguda la concentración de Malonil CoA en el músculo esquelético de ratas. Los modelos con animales indican que cuando el músculo es abastecido con combustibles extras que no son ácidos grasos, los niveles de Malonil CoA se incrementan (28, 107, 124, 125). A la inversa, los niveles de Malonil CoA decrecen cuando se priva al músculo de combustible, o el uso de energía se incrementa por contracción (28, 107, 124, 125).
Sin embargo, todos estos hallazgos son en modelos animales y no se hicieron mediciones directas de oxidación de ácidos grasos, o de algún aspecto del metabolismo de las grasas. Recientemente, reportamos que el flujo glucolítico incrementado durante el ejerció, a partir de hiperglucemia e hiperinsulinemia, redujeron la oxidación de ácidos grasos de cadena larga, pero no la de los de cadena media (25). Esto se interpretó para sugerir que el flujo glucolítico incrementado reduce activamente la entrada de ácidos grasos de cadena larga en la mitocondria, debido a que el transporte de los mismos a través de la membrana mitocondrial parece ser menos dependiente de la CAT I (106). Se sugirió que el flujo glucolítico incrementado puede haber aumentado la concentración de Malonil CoA en el músculo, tal como fue demostrado por Elayan y Zinder (31) lo cual puede haber inhibido la CAT I, y por lo tanto la entrada de ácidos grasos de cadena larga dentro de la mitocondria. Hallazgos similares fueron reportados por Sidossis et al (111), quienes provocaron el incremento en el flujo glucolítico aumentando la intensidad de ejercicio desde el 40 % hasta el 80 % del VO2 máx, observando una inhibición específica de la oxidación de ácidos grasos de cadena larga, en comparación con los de cadena media, a altas intensidades de ejercicio. Otra vez estos resultados sugieren que el trasporte de ácidos grasos de cadena larga dentro de la mitocondria es inhibido a nivel de la CAT I, lo cual puede estar regulado a través de la Malonil CoA (111). Si bien esta es una hipótesis atractiva, la misma puede no tener evidencia directa de los mecanismos mediadores de parte de la Malonil CoA, dado que las concentraciones de Malonil CoA en el músculo no fueron medidas en estos estudios (25, 111). Odland et al (90) fueron los primeros en medir Malonil CoA en el músculo esquelético humano. Ellos reportaron bajos niveles en reposo (comparado con los valores reportados en músculo esquelético de ratas), y una declinación del 20 % durante el ejercicio, aunque fallaron en demostrar significación estadística (90). Por ello, los primeros datos en humanos no son muy concluyentes y se necesita investigación adicional para dilucidar el rol de la Malonil CoA en la regulación de la oxidación de grasas. Otro problema no resuelto es que el Ki de la Malonil CoA para CAT I, medido “in vitro” es mucho mas bajo que la concentración medida en músculos de ratas y humanos “in vivo”, ambos en reposo y durante el ejercicio (84). De hecho a la concentración presente “in vivo”, se podría esperar una inhibición completa de la CAT I, pero esto no parece prevenir que los ácidos grasos con el tiempo se tornen un combustible incrementadamente importante en intensidades de ejercicio de suaves a moderadas. Una posibilidad es la concentración observada de la CAT I en la membrana mitocondrial externa, que de hecho es mucho mas baja que la concentración de Malonil CoA medida en el extracto del músculo entero, pero tampoco se puede excluir que el mecanismo de la Malonil CoA no sea tan importante como se sugiere en literatura reciente relacionada con la fisiología del ejercicio. Además, la concentración de Malonil CoA aún tiene que estar directamente vinculada a cambios en la oxidación de grasas durante el ejercicio en músculos de humanos o de ratas (122).
EL EFECTO DE LA INTENSIDAD DE EJERCICIO SOBRE EL METABOLISMO DE LAS GRASAS
La intensidad de ejercicio es el principal factor determinante del grado de oxidación de grasas o carbohidratos durante el ejercicio. En forma relativa, los ácidos grasos serán más importantes durante el ejercicio de baja intensidad. Durante el ejercicio al 25 % VO2 máx casi todo el gasto de energía derivó de las grasas (102). Durante el ejercicio al 65 % VO2 máx la oxidación de grasas aportó el 50 % del gasto de energía, pero dado que la renovación (“turnover”) de energía fue mucho más alta, las tasas absolutas de oxidación de grasas fueron mayores. Las tasas absolutas de provisión de energía de las grasas parece ser óptimo a niveles de ejercicio de entre 50 y 70 % VO2 máx. Romijn et al (102) investigaron el consumo de ácidos grasos plasmáticos y la utilización estimada de TGIM, utilizando isótopos estables. Ellos descubrieron que durante el ejercicio de baja intensidad (25 % VO2 máx) los TGIM contribuyen minimamente a la provisión de energía. Los ácidos grasos y la glucosa en plasma parecen ser los sustratos más importantes a esa intensidad donde las grasas son por lejos el combustible predominante. A intensidad de ejercicio moderada (65 % VO2 máx) los sustratos en el músculo (TGIM y glucógeno) se tornan mas importantes (Figura 3). Los TGIM fueron oxidados a altas tasas, a esta intensidad de ejercicio, mientras que los ácidos grasos plasmáticos fueron utilizados a una tasa levemente mas baja, comparado con el ejercicio de baja intensidad (Figura 3). Cuando la intensidad del ejercicio fue incrementada adicionalmente al 85% VO2 máx, la contribución de los ácidos grasos plasmáticos se tornó aun menor, mientras que también la oxidación de TGIM se redujo. La contribución decreciente de los ácidos grasos plasmáticos que se observó en este estudio, puede ser causada por una disminución en la disponibilidad de ácidos grasos, lo cual en cambio puede ser causado por tasas mas bajas de aparición de ácidos grasos en el plasma (TaAG) (71, 72). Esta TaAG disminuida (71), sin una reducción simultánea de la lipólisis (102) puede indicar que los ácidos grasos pueden quedar atrapados dentro del adipocito (56), tal vez como resultado del incremento de las concentraciones de lactato (67) o como resultado de la vasoconstricción en el tejido adiposo (12, 13, 104). Sin embargo. Aunque la movilización de ácidos grasos desde el tejido adiposo es uno de los factores que puede ser parcialmente responsable de la reducción en la oxidación de ácidos grasos durante el ejercicio a altas intensidades, esto puede no ser el único factor. Cuando los niveles de ácidos grasos en plasma estuvieron elevados a altos niveles, durante el ejercicio al 85 % VO2 máx, por infusión de “Intralipid” y heparina, las tasas de oxidación de grasas, en cierta forma se incrementaron (103), pero siguieron siendo más bajas que durante el ejercicio al 65 % VO2 máx (102). Esto indica que otros factores (intramusculares) pueden reducir la oxidación de ácidos grasos durante el ejercicio de alta intensidad. Un posible mecanismo puede ser que las altas tasas de glucólisis y altas tasas de formación de acetil CoA desde la glucosa-6-fosfato, a esas intensidades de ejercicio, inhiben el transporte de ácidos grasos de cadena larga dentro de la mitocondria al nivel de CAT I, incrementando las concentraciones de Malonil CoA (111). Además, durante el ejercicio intenso, serán reclutadas más fibras rápidas (FT) y menos fibras lentas (ST) (110). Dado que las fibras FT tienen una menor capacidad de oxidar ácidos grasos, la oxidación de grasas decrecerá, y concomitantemente, la oxidación de carbohidratos se incrementará cuando más de estas fibras sean reclutadas (110). También se sugirió que durante el ejercicio de alta intensidad hay un incremento en la competencia entre el piruvato que se convierte en acetil CoA vs los ácidos grasos que se transforman en acetil CoA, en una disputa por entrar dentro del ciclo ATC (60).
La contribución de los ácidos grasos se puede incrementar marcadamente cuando los depósitos de glucógeno en el músculo comienzan a repletarse (1). Esto implica, sin embargo, que la “alta” intensidad de ejercicio no puede ser sostenida, y que el ejercicio tiene que continuar a un nivel más bajo (92) debido a que la tasa de producción de ATP decrecerá.
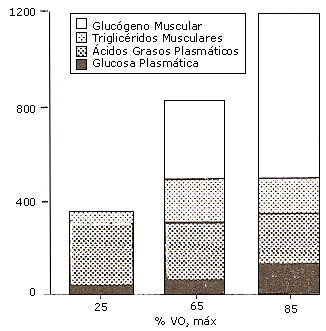
Figura 3. Utilización de sustratos a diferentes intensidades de ejercicio (25
% del VO2 máx, 65 % del VO2 máx y 85 % del VO2 máx). Datos adaptados de Romijn et
al (102).
ENTRENAMIENTO Y OXIDACION DE ACIDOS GRASOS
El entrenamiento de la resistencia afecta tanto la utilización de sustratos como la capacidad de ejercicio. Un gran numero de estudios, ambos en animales y hombres, establecieron un marcado incremento adaptativo en el potencial oxidativo en respuesta a una actividad física incrementada (57, 58). Una consecuencia notable, y probablemente un factor contribuyente a la capacidad de ejercicio mejorada luego del entrenamiento de resistencia, es el cambio del metabolismo hacia un mayor uso de las grasas y un concominante ahorro de los depósitos de glucógeno (6, 18, 46, 47, 57-59, 64, 69, 109, 110). El incremento en el rendimiento y el cambio hacia el metabolismo de grasas durante el ejercicio sub-máximo son más pronunciados que el cambio en el máximo consumo de oxígeno corporal total (% VO2 máx) (45), y parece improbable que otras adaptaciones al entrenamiento como un incremento en el volumen minuto cardíaco máximo o en la función pulmonar sean un factor principal para explicar el cambio del metabolismo de carbohidratos al de grasas en el músculo esquelético entrenado (47). La contribución de las grasas al gasto total de energía se incrementa luego del entrenamiento, tanto a la misma intensidad relativa de ejercicio como a la absoluta (23, 54, 58, 64, 68, 69). Esto es de suma importancia durante el ejercicio prolongado de moderada a alta intensidad (50-90 % VO2 máx) dado que se requieren carbohidratos para mantener esos niveles de ejercicio. Tan pronto como los depósitos de glucógeno se comienzan a depletar y la oxidación de carbohidratos cae por debajo de un nivel crítico, la intensidad de ejercicio tiene que ser reducida, ya que los ATP no pueden ser generados a una tasa suficientemente alta (87).
Si bien las ventajas de la oxidación de grasas incrementada durante el ejercicio son obvias, los mecanismos celulares y moleculares que se refieren a este efecto benéfico del entrenamiento, son entendidos en forma incompleta. Diversas adaptaciones pueden contribuir a una estimulación de la oxidación de grasas en sujetos entrenados: 1) un incremento en el número de enzimas oxidativas, y del contenido mitocondrial en el músculo entrenado; 2) un aumento en la oxidación de triacilglicéridos en el músculo; 3) un incremento en el consumo o captación celular de ácidos grasos; 4) alteraciones en la movilización de ácidos grasos desde el tejido adiposo. No solo la causa de la oxidación incrementada de ácidos grasos luego del entrenamiento es incierta, sino también la fuente o procedencia de estos ácidos grasos sigue sin ser completamente dilucidada.
Un Incremento en el Número de Enzimas Oxidativas y en el Contenido Mitocondrial
Estudios acerca de la homogeneidad de todo el músculo demostraron que el músculo esquelético de las ratas efectúa incrementos adaptativos en las capacidades para oxidar ácidos grasos (6, 86) y cetonas (126). Basados en la cinética de las enzimas, Gollnick y Saltin (46) calcularon que la mejoría en la actividad de las enzimas involucradas en la oxidación de ácidos grasos puede ser la primera causa del incremento de la oxidación de grasas, luego del entrenamiento. Frecuentemente, se han reportado niveles incrementados de las enzimas involucradas en la activación del transporte y en la beta- oxidación de los ácidos grasos de cadena larga (6, 15, 24, 57, 86). Se hallaron niveles aumentados de 3-hidroxiacil CoA-dehidrogenasa en ratas entrenadas en resistencia (27), y en el hombre (64, 68). Otras enzimas que sostienen el metabolismo de los ácidos grasos, las cuales tienen un incremento de su actividad o de su contenido luego del entrenamiento, incluyen: carnitin palmitoil-transferasaI (186), carnitin acil-transferasa (86) y acil CoA graso-sintetasa (86). Sin embargo, no solo las enzimas involucradas en la activación, transporte y oxidación de ácidos grasos se incrementan en el músculo entrenado, sino también las enzimas involucradas en el ciclo ATC y en la cadena respiratoria (15, 24, 54). Dado que el entrenamiento de la resistencia incrementa la capacidad de oxidar ácidos grasos como también de piruvato, subsiste la pregunta: ¿por qué son oxidados proporcionalmente más ácidos grasos y menos carbohidratos? Esto puede ser explicado por la densidad mitocondrial, como lo discutieron Gollnick y Saltin (46).
El entrenamiento de la resistencia incrementa tanto el tamaño como el número de mitocondrias (58). Esto incrementa el área de superficie donde puede tener lugar el intercambio de sustratos y el ADP, y posiblemente el numero de proteínas de transporte. Gollnick y Saltin (46) propusieron que el volumen mitocondrial total aumentado, tal como se observa luego del entrenamiento, incrementa la capacidad de transportar ADP formado por el ciclo contráctil dentro de la mitocondria. Consecuentemente, lo niveles de ADP libre (ADP l) son mas bajos en los músculos entrenados en el ejercicio que en los músculos de no entrenados, a la misma actividad contráctil (22). Se demostró que la concentración de ADP l, el cociente ATP/ADP l, y el cociente ATP/(ADP l x Pi), tanto en el citoplasma como en la mitocondria, son factores regulatorios “llaves” del metabolismo (4, 29, 114). Al margen de estos factores, también el Pi extra-mitocondrial y el suministro de hidrogeno fueron propuestos como importantes factores regulatorios de la respiración mitocondrial (114). De acuerdo al modelo de Gollnick y Saltin (46), manteniendo niveles mas bajos de ADP l, o incrementos en el cociente ATP/ADP l, se favorecería una mayor entrada a las vías oxidativas de acetil CoA provista por la degradación de ácidos grasos, debido a que el ADP l y el cociente ATP/ADP l tienen una influencia estimulante sobre la glucólisis (88).
Comparados con no entenados, los sujetos entrenados tienen un contenido mitocondrial más alto (e.g., mayor capacidad oxidativa), y durante el ejercicio a cierta carga de trabajo se formará menos ADP l [junto con cocientes ATP/ADP l y ATP/(ADP l x Pi) mas altos]. Estos cambios controlarán directamente el metabolismo de energía estimulando la glucógeno-fosforilasa, la PFK y la PDH, lo que resultará en un incremento del flujo glucolítico.
Si bien la actividad de las enzimas y la densidad mitocondrial se incrementan luego del entrenamiento, queda por determinar si la actividad de las enzimas es la limitación principal en a oxidación de lípidos en la célula del músculo en ejercicio. Un incremento en la capacidad oxidativa debería estar acompañado de un aumento cuantitativamente similar en el suministro de ácidos grasos a la mitocondria (119). La evidencia para sostener esta visión es la observación de que los corredores con tasas similares de oxidación de grasas, durante una carera de 60 min al 70 % VO2 máx, presentaron diferencias considerables en la actividad de la carnitín palmitoil-transferasa I y de la succinil-dehidrogenasa (24).
Malonil CoA y Utilización de Grasas Luego del Entrenamiento
Tal como se discutió previamente, la Malonil CoA puede ser un regulador importante de la oxidación de ácidos grasos en el músculo esquelético durante el ejercicio (124, 125). Niveles reducidos de Malonil CoA durante el ejercicio pueden permitir que más ácidos grasos de cadena larga sean transportados dentro de la matriz mitocondrial, dado que la CAT 1 es menos inhibida. Las concentraciones disminuidas de Malonil CoA en músculo durante el ejercicio son paralelas a la reducción en el flujo de carbohidratos. Recientemente se ha provisto evidencia de que el flujo de carbohidratos regula directamente la oxidación de ácidos grasos, durante el ejercicio al 50 % VO2 máx (23, 112). Por lo tanto, es tentador especular que el entrenamiento puede resultar en una mayor caída de las concentraciones de Malonil CoA durante el ejercicio, mitigando por lo tanto la inhibición de CAT I, y mejorando la oxidación de ácidos grasos. Sin embargo, estos mecanismos son más bien especulativos y se necesita investigación adicional para evaluar estas hipótesis contradictorias.
El entrenamiento también reduce el consumo de glucosa durante el ejercicio a la misma intensidad absoluta de ejercicio (18, 20), aunque el numero de transportadores de glucosa (GLUT 4) en el músculo se puede incrementar (52). Se ha sugerido que esta baja en el consumo de glucosa plasmática, en combinación con un incremento en a oxidación de grasas luego del entrenamiento, es regulada a través del clásico ciclo glucosa-ácidos grasos. Sin embargo, Coggan et al (20) reportaron que las concentraciones de citrato y de glucosa-6-fosfato fueron más bajas en el estado entrenado que en el no entrenado, a la misma intensidad de ejercicio, lo cual contrasta con el concepto del ciclo glucosa-ácidos grasos.
Efecto del Entrenamiento sobre la Utilización de Ácidos Grasos Plasmáticos
Siendo el consumo y la oxidación de ácidos grasos dependiente de su concentración vascular y del potencial oxidativo incrementado del músculo esquelético entenado en resistencia (2, 53, 65, 73, 116), se podría esperar que una mejor tasa de lipólisis acompañe el entrenamiento. Esto puede ser representado como un mecanismo para proveer más ácidos grasos al músculo activo, para dar sustento al potencial incrementado para oxidar ácidos grasos. Sin embargo, concentraciones mas bajas de ácidos grasos en plasma luego del entrenamiento (26, 64, 81, 127) sugieren que el efecto del entrenamiento sobre la oxidación de ácidos grasos plasmáticos, durante el ejercicio, es por una disminución de la movilización desde el tejido adiposo, o por un incremento en la extracción de los ácidos grasos por parte del músculo.
Un estudio de Martin et al (81) utilizando trazadores isotópicos de palmitato ha demostrado que tanto la tasa de aparición (Ta) como la tasa de desaparición (Td) de ácidos grasos están disminuidas como resultado del entrenamiento. Henriksson (54) descubrió tasas similares de consumo de ácidos grasos entre una pierna entrenada y una no entrenada, durante el ejercicio de dos piernas de 50 min a moderada intensidad; y Janson y Kaijser (68) no hallaron efecto alguno del entrenamiento sobre la extracción de ácidos grasos plasmáticos en las piernas. Por lo tanto, si bien la oxidación de grasas está marcadamente aumentada, es improbable que los ácidos grasos plasmáticos sean la fuente principal de ácidos grasos que explican este incremento en la oxidación. Los estudios mencionados previamente (54, 64, 68, 81) respaldan el concepto de que el entrenamiento no incrementa la extracción de ácidos grasos plasmáticos por parte del músculo. Sin embargo, Turcotte et al (117) reportaron que, por encima de ciertos niveles de ácidos grasos en plasma, el consumo muscular de ácidos grasos plasmáticos fue limitado en sujetos no entrenados, en comparación con los sujetos entrenados, durante la tercer hora de un ejercicio de extensión de rodilla al 60 % VO2 máx. Mientras que el consumo de ácidos grasos plasmáticos se elevó linealmente con el incremento en las concentraciones de ácidos grasos (no unidos a proteínas) en sujetos entrenados, el mismo siguió la cinética de saturación en sujetos no entrenados con concentraciones en plasma por encima de 700 mmol/L. La observación de que el consumo de ácidos grasos es un proceso saturable fue demostrada previamente en un modelo en ratas (116). El entrenamiento puede provocar cambios en el trasporte a través de la membrana (117). Se sugirió que las PUAG, TAG y TAGP pueden desempeñar un rol importante en el transporte a través de la membrana y dentro del citoplasma (44, 119). En ratas, sin embargo, el entrenamiento incrementó el contenido de PUAG del músculo cardiaco, pero no en el Extensor Digitorium Longus (EDL) y en el sóleo (118). Sin embargo, como fue discutido por Coggan (21), los resultados de estudios que utilizaron ejercicios de extensión de rodilla, como los que aplicaron Turcotte et al (117), no pueden ser fácilmente extrapolados al ejercicio de todo el cuerpo dado que con este ejercicio de extensión de rodilla los cambios hormonales son mucho mas pequeños, comparados con el ejercicio corporal total. Dado que la extracción de ácidos grasos no parece incrementarse luego del entrenamiento durante el ejercicio corporal total (68, 81), la reducción de la oxidación de ácidos grasos plasmáticos puede deberse a una lipólisis reducida. La explicación más probable para un efecto de entrenamiento sobre la lipólisis del tejido adiposo es, probablemente, un detrimento en la respuesta simpato-adrenal a un ejercicio de la misma intensidad absoluta. El nivel de actividad simpato-adrenal, el cual es el principal determinante de la lipólisis del tejido adiposo en humanos, es marcadamente mitigado aún luego de varias semanas de entrenamiento (127). Zinder et al (127) observaron una reducción del 55 % en las concentraciones plasmáticas de catecolaminas durante el ejercicio prolongado (la frecuencia cardiaca reducida puede ser un indicador de actividad simpato-adrenal disminuida luego del entrenamiento). El entrenamiento de la resistencia también atenúa el incremento inducido por el ejercicio de las concentraciones en plasma de otras hormonas lipolíticas, tal como el glucagón y la hormona de crecimiento (STH) (127). Asimismo, Lavoie et al (78) demostraron que luego del entrenamiento de resistencia se incrementa el efecto inhibidor de la insulina sobre la lipólisis, lo cual conduce a una menor liberación (Ta) de ácidos grasos (78, 81), y a una reducción en las concentraciones de ácidos grasos plasmáticos (18, 78, 81, 127). Sin embargo, si bien la reducción de la liberación de ácidos grasos luego del entrenamiento puede principalmente ser explicada por la tasa de lipólisis reducida, no se puede excluir el hecho que los sujetos entrenados tienen tasas más altas del ciclo triacilglicéridos-ácidos grasos. Se reportó que la lipólisis disminuida (medida por Ta de glicerol) a la misma intensidad absoluta de ejercicio es la misma (75), o es levemente menor (93) luego del entrenamiento, mientras que la lipólisis corporal total puede incrementarse a la misma intensidad de ejercicio relativa (76).
Interesantemente, la lipólisis en el tejido adiposo parece estar reducida durante el entrenamiento, mientras que al mismo tiempo la lipólisis en el músculo esquelético parece estar incrementada (ver “efectos del entrenamiento sobre la utilización de TGIM”). El mecanismo que está detrás de estos efectos desiguales del entrenamiento no es claro. Sin embargo. Existen varias diferencias en la lipólisis en el músculo esquelético y el tejido adiposo que pueden, al menos, explicar parcialmente las diferentes respuestas al entrenamiento. Ante todo, la lipólisis del músculo esquelético parece ser mas sensible al bloqueo beta-adrenérgico que la lipólisis del tejido adiposo (16), lo que implica que, a niveles mas bajos de estimulación simpato-adrenal (tal como luego del entrenamiento [127]), la lipólisis será relativamente mas estimulada en el músculo esquelético. En segundo lugar, se ha sugerido que existe una sub-regulación de la lipólisis específica del tejido en el estado entrenado (81), tal como lo evidenció la actividad incrementada de la adenilato-ciclasa en el músculo esquelético de ratas entrenadas, comparadas con no entrenadas (10). Tampoco se puede excluir que el ejercicio conduce a la activación alostérica de la LHS, especialmente en músculos entrenados, o que el entrenamiento conlleva a un incremento en la concentración de la enzima LHS en el músculo. Sin embargo, solo podemos especular acerca de esta interesante diferencia entre individuos entrenados y no entrenados, ya que el mecanismo exacto no se conoce hasta el momento.
Efecto del Entrenamiento sobre la Utilización de TGIM
La oxidación de ácidos grasos plasmáticos a pesar de las tasas más altas de oxidación total de grasas luego del entrenamiento sugieren que la grasa adicional oxidada durante el ejercicio tiene que derivar de otras fuentes que no sean los triacilglicéridos del tejido adiposo, y posiblemente de los triacilglicéridos del músculo (53, 64, 66, 81). Algunos (73, 81), pero no todos (64) los estudios reportaron que los músculos esqueléticos entrenados incrementaron las concentraciones de triacilglicéridos musculares, lo cual puede servir como una fuente de energía significativa durante el ejercicio (14, 32, 33, 37, 64, 68, 97, 113). Debido a que las gotitas de lípidos están ubicadas en cercana proximidad a la mitocondria (61), el tiempo de tránsito de los ácidos grasos desde los TG en una gotita de lípidos intramuscular hacia la membrana mitocondrial externa será muy breve. Por lo tanto, también desde un punto de vista teleológico, el incremento en los depósitos de triacilglicéridos musculares podría tener una ventaja práctica. La observación de que el entrenamiento incrementa el número de gotitas de lípidos (62) se alinea con el reporte de un incremento de la oxidación de triacilglicéridos intramusculares luego del entrenamiento (53, 64,66, 81, 93). De alguna manera paradójica, el entrenamiento reduce el “drive” adrenérgico lo cual provoca la reducción de la lipólisis en el tejido adiposo (93), pero incrementa la lipólisis intramuscular y la oxidación de ácidos grasos (ver discusión previa: “efecto del entrenamiento sobre la utilización de ácidos grasos en plasma”). Falta evidencia del efecto directo del entrenamiento sobre la actividad de la LHS en el músculo.
En resumen, se ha demostrado consistentemente que el entrenamiento incrementa el “pool” de TGIM y su oxidación durante el ejercicio, a la misma intensidad absoluta. El mecanismo por el cual el ejercicio incrementa la utilización de TGIM no está bien comprendido.
Efecto del Entrenamiento sobre la Utilización de VLDL-TG Plasmáticos
Es tentador creer que la actividad de la LPL (3, 74, 89) y una mayor área de superficie capilar endotelial luego del entrenamiento (74), serán responsables del incremento de la lipólisis de triacilglicéridos plasmáticos, luego del entrenamiento. Se ha demostrado que los incrementos en la actividad de la LPL están linealmente relacionados a incrementos en la capilarización del músculo entrenado (74). Si bien las tasas de extracción de los TG circulantes fueron relativamente pequeñas e inconsistentes (74). Kiens y Lithel (74) sugirieron, por lo tanto, que la degradación de VLDL-Triacilglicérido probablemente sea más importante para su potencial influencia a largo plazo sobre los perfiles lipídicos en sangre, que la contribución para una tasa más alta de oxidación de grasas durante el ejercicio luego del entrenamiento. Además, el entrenamiento parece disminuir más que incrementar la disponibilidad de VLDL circulante (50), reduciendo la producción de las mismas por parte del hígado.
CONCLUSIONES
En conclusión, la densidad mitocondrial incrementada luego del entrenamiento y el aumento de las enzimas oxidativas pueden explicar, parcialmente, el incremento de la oxidación de ácidos grasos durante el ejercicio, tal como se observó después de un periodo de entrenamiento. Sin embargo, también el suministro de ácidos grasos a la mitocondria puede ser importante. La evidencia disponible sugiere que los de ácidos grasos oxidados en forma adicional, luego del entrenamiento, son principalmente derivados de triacilglicéridos intramusculares y no provenientes de ácidos grasos derivados del tejido adiposo o de triacilglicéridos circulantes.
Referencias
1. Ahlbor G. Bjorkman O (1987). Carbohydrate utilization by exercising muscle following pre exercise glucose ingestion. Clin Physiol 7: 181-95
2. Armstrong DT. Steele R. Altszuler N. Dunn A. Bishop JS. De Bodo RC (1961). Regulation of plasma free fatty acid turnover. Am J Physiol 201: 9-15
3. Baghy GJ, Johnson Jl. Bennett BW. Shepherd RE (1986). Muscle lipoprotein lipase activity in voluntary exercising rats. J Appl Physiol 60: 1623-7
4. Balaban RS (1990). Regulation of oxidative phosphorylation in the mammalian cell. Am J Physiol 258: C377-89
5. Balasse EO, Neef MA (1974). Operation of the glucose-fatty acid cycle during experimental elevations of plasma free fatty acid levels in man. Eur J Clin Invest 4: 247-52
6. Baldwin KM. klinkerfuss GH, Terjung RI. Mole PA, Holloszy JO (1972). Respiratory capacity of white, red, and intermediate muscle adaptative response to exercise. Am J Physiol 222: 373-8
7. Baron AD, Brechtel G. Edelman SV (1989). Effects of free fatty acids and ketone bodies on in vivo non-insulin-mediated glucose utilization and production in humans. Metabolism 38: 1056-61
8. Berger M. Hagg SA. Goodman MN. Ruderman NB (1976). Glucose metabolism in perfused skeletal muscle. Bioch J 1976: 158: 191-202
9. Bergstrom J. Hultman E. Jorfeldt I., Pernow B, Wahren J (1969). Effect of nicotinic acid on physical working capacity and on metabolism of muscle glycongen in man. J Appl Physiol 26
10. Buckenmeyer PJ. Goldfarb AH, Partilla JS. Pineyro MA, Dax EM (1990). Endurance training, not acute exercise, differentially alters β-receptors and cyclase in skeletal muscle fibres. Am J Physiol 258: E71-7
11. Bulow J (1982). Subcutaneous adipose tissue blood flow and triacylglycerol-mobilization during prolonged exercise in dogs. Pfluglers Arch 392: 230-4
12. Bulow J (1983). Adipose tissue blood flow during exercise. Dan Med Bull 30: 85-100
13. Bulow J. Madsen J (1978). Adipose tissue blood flow during prolonged heavy exercise II. Pflugers Arch 378: 41-5
14. Chi MM-Y, Hintz CS, Coyle EF. Martin III WH, Ivy JI., Nemeth PM, Holloszy JO, Lowry OH (1983). Effects of detraining on enzymes of energy metabolism in individual human muscle fibers. Am J Physiol 244: C276-87
15. Cleroux J. van Nguyen P. Taylor AW. Leenen FHH (1989). of β1 vs. β1 + β2-blockade in exercise endurance and muscle metabolism in humans. Effects. J Appl Physiol 66: 548-54
16. Coakley JH, Wagenmakers AJM. Edwards RHT (1992). Relationship between ammnia, heart rate and exertion in McArdle is disease. Am J Physiol 262: E167-72
17. Coggan AR. Kohrt WM. Spina RJ. Bier DM. Holloszy JO (1990). Endurance training decreases plasma glucose turnover and oxidation durin moderate-intensity exercise in man. J Appl Physiol 68: 990-6
18. Coggan AR. Mendenhall LA (1992). Effect of diet on substrate metabolism during exercise. Energy metabolism in exercise and sport. Dubuque Bronw & Benchmark. 435-71
19. Coggan AR. Spina RJ. Kohrt WM. Holloszy JO (1993). Effect of prolonged exercise on muscle citrate concentration before and after endurance training in men. J Appl Physiol 264: E215-20
20. Coggan AR. Williams BD (1995). Metabolic adaptations to endurance training substrate metabolism during exercise. Exercise metabolism. Champaign. Human Kinetics Publishers. 41-71
21. Constable SH. Favier RJ. Mr McLane JA. Fell RD, Chen M. Holloszy JO (1987). Energy metabolism in contracting rat skeletal muscle adaptations to exercise training. Am Appl Physiol 253: C316-C322
22. Costill DL. Coyle E. Dalsky G. Evans W. Fink W, Hoopes D (1977). Effects of elevated plasma FFA and insulin on muscle glycogen usage during exercise. J Appl Physiol 43: 695-9
23. Costill DL. Fink WJ. Getchell LH, Ivy Jl, Witzman FA (1979). Lipid metabolism in skeletal muscle of endurance trained males and females. J Appl Physiol 47: 787-91
24. Coyle EF. Jeukendrup AF. Wagenmakers AJM, Saris WHM (1997). fatty acid oxidation is directly regulated by carbohydrate metabolism during exercise. Am J Physiol 273 in press
25. Datsky G. Martin W. Hurley B. Matthews D. Bier, D. Hahberg J. Holloszy JO (1988). Oxidation of plasma FFA during endurance exercise. Med Sci Sports Exerc 16: 202
26. Degens H. Veerkamp JH. Van Moekerk HTR. Furek Z. Hoofd LJC. Bunkhorst RA (1993). Metabolic capacity, fibre type area and capillarization of rat plantaris muscle. Effects of age, overload and training and relationship with fatigue resistance . Int. J. Biochem. 25: 9109-14
27. Duan C. Winder WW (1992). Nerve stimulation decreases malonyl-CoA in muscle. J Appl Physiol 72: 901-4
28. Dudley GA. Tullson PC. Ferjung RL (1987). Influence of mitochondrial content on the sensivity of respiratory control. J Biol Chem 262: 9109-14
29. Dyck DJ, Putman CT. Heigenhauser GJF. Hultman E. Spriet LI (1993). Regulation of fat-carbohydrate interaction in skeletal muscle during intense aerobic cycling. Am J Physiol 265: E852-9
30. Elavan IM. Winder WW (1991). Effect of glucose infusion on muscle malonyl-CoA during exercise. Appl Physiol 70: 1495-9
31. Essen B (1940). Intramuscular substrate utilization during pronged exercise. Ann N TAcad Sci. New York
32. Essen Gustavsson (1977). during pronged exercise. Ann N Y Acad Sci. New York: New York Academy of Sciences 30-44
33. Farrel PA (1992). Exercise effects on regulation of energy metabolism by pancreatic and gut hormones. Energy metabolism in exercise and sport. Dubuque, Brown & Benchmark. 53-106
34. Felber JP. Vannotti A (1994). Effects of fat infusion on glucose tolerance and insulin plasma levels. Med Exp 10: 153-6
35. Ferrannini E. Barrett EJ. Bevilacqua S. DeFronzo RA (1983). Effect of fatty acids on glucose production and utilization in mans. J Clin Invest 72: 1737-47
36. Galbo H (1981). Endocrinology and metabolism in exercise. Int. J Sports Med 2: 203-11
37. Galbo H (1983). Hormonal and metabolic adaptation to exercise. Stuttgart. Thieme
38. Galbo H. Holts JJ. Christensen NJ (1979). The effect of different diets and of insulin on the hormonal response to prolonged exercise. Acta Physiol Scand 107: 19-32
39. Galbo H. Richter EA, Holts JJ. Christensen NJ (1977). Diminished hormonal responses to exercise in trained rats. J Appl Physiol 43: 953-8
40. Garland PB, Newsholme EA. Randle PJ (1964). Regulation of glucose uptake by muscle: effect of fatty acids and ketone bodies, and of alloxan-diabetes and starvation, on pyruvate metabolism and on lactate pyruvate and L-glycerol 3-phosphate-dihydroxycetone . Biochem J 93: 665-78
41. Garland PB, Randle Pj (1994). Regulation of gucose uptake by muscle: effects of alloxan diabetes, starvation, hypophysectomy and adrenalectomy, and of fatty acids, ketone bodies and pyruvate on the glycerol output and concentrations of free fatty acids. A. glycerol phosphate and citrate cycle intermediates in rat heart and diaphragm muscles. Biochem J 93: 678-87
42. Glatz JFC, van der Vusse GJ. Veerkamp JH (1988). Fatty acid binding proteins and their physiological significance. NIPS 3: 41-3
43. Gollnick PD (1985). Metabolism if substrates, energy substrate metabolism during exercise and as modified by training. Fed Proc 44: 353-7
44. Gollnick PD. Saltin B (1982). Significance of skeletal muscle oxidative enzyme enhancement with endurance training. Clin Phys 2: 1-12
45. Gollnick PD. Saltin B (1988). Fuel for muscular exercise. Exercise, nutrition and energy metabolism. New York: Macmillan Publishing Company. 71-88
46. Goodman HM (1970). Permissive effects of hormones on lipolysis. Endocrinology 86: 1064-74
47. Goodman MN. Berger M Ruderman NB (1974). Glucose metabolism in rat skeletal muscle at rest. Diabetes 23: 881-8
48. Gorski J. Oscai J.B. Palmer WK (1990). Hepatic lipid metabolism in exercise and training. Med Sci Sports Exerc 22: 213-21
49. Hargreaves M. Kiens B. Richter EA (1991). Effect of increased plasma free fatty acid concentrations on muscle metabolism in exercising men. J Appl Physiol 70: 194-201
50. Hargreaves M. McCoy ;. McConell G. Proietto J (1994). Skeletal muscle GLUT4 and glucose uptake during exercise in humans. Clin Sci 87: 67
51. Havel RJ. Pernow B. Jones NI (1967). Uptake and release of free fatty acids and other metabolitos in the legs of exercising men. J Appl Physiol 23: 90-9
52. Enriksson J (1977). Training-induced adaptation of skeletal muscle and metabolism during submaximal exercise. J Physiol 270: 661-75
53. JHickson RC. Rennie MJ. Conlee RK. Winder WW. Holloszy JO (1977). Effect of increased plasma fatty acids on glycogen untilization and endurance. J Appl Physiol 43: 829-33
54. Hodgetts V. Coppack SW. Frayn KN Hockaday TDR (1991). Factors controlling fat mobilization from human subcutaneous adipose tissue during exercise. J Appl Physiol 71: 445-51
55. Holloszy JO. Booth W (1976). Biochemical adaptations to endurance exercise in muscle. Ann Rev Physiol 38: 273-91
56. Holloszy JO. Coyle EF (1984). Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. Appl Physiol 56: 831-8
57. Holloszy JO. Dalsky GP, Nemeth PM, Hurley BF, Martin III WH, Hagberg JM (1986). Utilization of fat as substrate during exercise: effect of training. Biochemistry of exercise VI. Illinois: Hum in Publ. 183-90
58. Holloszy JO. Kohrt WM (1996). Regulation of carbohydrate and fat metabolism during and after exercise. Ann Rev Nutr 16: 121-38
59. Hoppeler H (1986). Exercise-induced ultrastructural changes in skeletal muscle. Int J Sport Med 7: 187-204
60. Hoppeler H. Howald H. Conley KE. Lindstedt SI., Claasem H. Vonck P. Weibel ER (1985). Endurance training humans: aerobic capacity and structure of skeletal muscle. J Appl Physiol 59: 320-7
61. Horowits JF. Mora Rodriguez R. Byerley I.O. Coyle EF (1987). Lipolytic supresión following, carbohydrate ingestión limits fat oxidation during exercise. Abstracts 4: 0145 F
62. Hurley BF Nmeth PM Martin III WH, Hagberg JM, Dalsky GP. Holloszy JO (1964). Muscle triglyceride utilization during exercise effect of training. J Appl Physiol 60: 562-7
63. Issekutz B. Bortz WM, Miller HI, Paul P (1967). Turnover rate of plasma FFA in humans and in dogs. Metabolism 16: 1001-9
64. Issekutz B. Miller HI, Paul P. Rodahl K (1964). Source of fat in exercising dogs. Am J Physiol 207: 583-9
65. Issekutz B. Shaw WA. Issekuts TB (1975). Effect of lactate on FFA and glycerol turnover in resting exercising dogs. J Appl Physiol 39: 349-53
66. Jansson E. Kaijser I (1987). Substrate utilization and enzymes in skeletal muscle of extremely endurance-trained men. J Appl Physiol 62: 999-1005
67. Jeukendrup AE. Mensink M. Saris WHM, Wagenmakers AJM (1997). Exogenous glucose oxidation during exercise in endurance-trained and untrained subjects. J Appl Physiol 82: 835-40
68. Jeukendrup AF. Saris WHM. Wagenmakers AJM (1998). Fat metabolism during exercise: a review. Part I. Fatty acid mobilization and muscle metabolism Int J Sports Med 19: 1-14
69. Jeukendrup AE. Saris WHM, Wagenmakers AJM (1998). Fat metabolism during exercise: a review. Part III: Effects of nutritional interventions. Int J Sports Med 19: in press
70. Jones NI. Helgenhauser GJF. Kuksis A. Matsos CG. Sutton JR, Toews CJ (1980). Fat metabolism in heavy exercise. Clin Sci 59: 469-78
71. Kanaley JA. Mottram CD. Scanlon PD, Jensen MD (1995). Fatty acid kinetic response to running above or below lactate threshold. J Appl Physiol 79: 439-47
72. Klens B. Essen-Gustavsson B. Christensen NJ. Saltin B (1993). Skeletal muscle substrate utilization during submaximal exercise in man: effect of endurance training. J Physiol 469: 459-78
73. Klens B. Lithell H (1989). Lipoprotein metabolism influenced by training induced changes in human skeletal muscle. J Clin Invest 83: 558-64
74. Klein S. Coyle EF. Wolfre RR (1994). Fat metabolism during low-intensity exercise in endurance-trained and untrained men. Am J Physiol 267: E934-40
75. Klein S. Weber J-M. Coyle EF. Wolfre RR (1996). Effect of endurance training on glycerol kinetics during strenuous exercise in humans. Metabolism 45: 357-61
76. Knapik JJ. Meredith CN. Jones BH. Suek , Young VR, Evans WJ (1988). Influence of fasting on carbohydrate and fat metabolism during rest and exercise in men. J Appl Physiol 64: 1923-9
77. Li J. Stiliman S. Clore JN, Blackard WG (1993). Skeletal muscle lipids and glycogen mask substrate competition (Randle Cycle). Metabolism 42: 451-6
78. Loy SF. Conlee RK. Winder WW. Nelson AG. Arnall DA. Fisher AG (1986). Effect of 24-hour fast on cycling endurance time at two different intensities. J Appl Physiol 61: 654-9
79. Martin III WH, Dalsky GP, Hurley BF. Matthews DF., Bier DM, Hagberg JM. Rogers MA, King DS. Holloszy JO (1993). Effect of endurance training on plasma free fatty acid turnover and oxidation during exercise. Am Physiol 265: E708-14
80. McGarry JD, Foster DW (1980). Regulation of hepatic fatty acid oxidation and ketone body production. Ann Rev Biochem 49: 395-420
81. McGarry JD, Mills SE, Long CS Foster DW (1983). Observations on the affinity for carnitine and malonyl-CoA sensivity of carnitine palmitoyl transferase I in animal and human tissues. Bioch J 214: 21-8
82. McGarry JD, Stark MJ. Foster DW (1978). Hepatic malonyl-CoA levels of red, fasted and diabetic rats as measured using simple radioisotopic assay. J Biol Chem 253: 8291-3
83. Newsholme EA (1989). Metabolic causes of fatigue in track events and the marathon. Advances in myochemistry. John Libbey Eurotext Ltd. 263-71
84. Newsholme EA, Leech AR (1990). Biochemistry for the medical sciences. Chichester: John Wiley & Sons
85. Odland LM, Heigenhauser GJF, Lopaschuk GD, Spriet LL (1996). Human skeletal muscle malonyl-CoA at rest and during prolonged submaximal exercise. Am J Physiol 270: E541-4
86. Pearce FJ. Connett RJ (1980). Effect of lactate and palmitate on substrate utilization of isolated rat soleus. Am J Physiol 238: C149-59
87. Pernow B. Saltin B (1971). of substrates and capacity for prolonged heavy exercise in man Availability. J Appl Physiol 31: 416-22
88. Phillips SM, Green HJ. Tarnopoolsky MA, Heigenhauser GF, Hill RE., Grant SM (1996). Effects of training duration on substrate turnover and oxidation during exercise. J Appl Physiol 81: 2182-91
89. Randle PJ. Hales CN, Garland PB, Newsholme EA (1963). The glucose-fatty acid cycle: its role in insulin sensitivity and the metabolic disturbances if diabetes mellitus. Lancet 1: 785-9
90. Randle PJ. Newsholme EA. Garland PB (1964). Regulation of fatty acids, ketone bodies and pyruvate, and of alloxan diabetes and starvation on the uptake and metabolic rate of glucose in rat heart and diaphragm muscles. Biochem J 93: 652-65
91. Ravussin E. Bogardus C. Scheidegger K. LaGrange B. Horton ED. Horton ES (1986). Effect of elevated FFA on carbohydrate and lipid oxidation during prolonged exercise in humans. J Appl Physiol 60: 893-900
92. Reitman J. Baldwin KM. Holloszy JO (1973). Intramuscular triglyceride utilization by red, white and intermediate skeletal muscle and heart during exhaustive exercise. 628-31
93. Rennie MJ. Holloszy JO (1977). Inhibition of glucose uptake and glycogenolysis by availability of oleate in well-oxygenated perfused skeletal muscle. Bioch J 168: 161-70
94. Rennie MJ. Winder WW (1940). A sparing effect of increased plasma fatty acids on mscle and liver glycogen content in the exercising rat. Bioch J 156: 647-55
95. Richter EA (1984). Influence of the sympatho-adrenal system on some metabolic and hormonal responses to exercise in the rat. Acta Physiol Scand suppl 528: 1-42
96. Richter EA. Ruderman NB, Gavras H. Belur ER. Galbo H (1982). Muscle glycogenolysis during exercise: dual control by epinephrine and contractions. Am J Physiol 242: E25-32
97. Romijn JA. Coyle EF. Sidossis LS. Gastaldelli A. Horowitz JF. Endert E. Wolfre RR (1993). Regulation of endogeneous fat and carbohydrate metabolism in relation to exercise intendity. Am J Physiol 265: E380-91
98. Romijn JA. Coyle EF. Sidossis LS. Zhang X-J. Wolfe RR (1995). Relationship between fatty acid delivery and fatty acid oxidation during strenuous exercise. J Appl Physiol 79: 1939-45
99. Rosell S. Belfrage E (1979). Blood circulation in adipose tissue. Phy Rev 59: 1078-104
100. Saddik M. Gamble J. Witters LA. Lopaschuk GD (1993). Acetyl-CoA carboxylase regulation of fatty acid oxidation in the heart. J Biol Chem 268: 25836-45
101. Saggerson ED. Carpenter CA (1981). Carnitine palmitoyltransferase and carnitine octanoyltransferase activites in liver, kidney cortex, adipocyte, lactating mammary gland, skeletal muscle and heart. FEBS left 129: 229-32
102. Saha AK, Kurowski TG (1995). Ruderman NB a malonyl-CoA mechanism in muscle: effect of insulin, glucose, and denervation. Am J Physiol 269: F283-9
103. Saha AK. Vavyas D. Kurowski TG, Apazidis A. Witters LA. Shafrir E. Ruderman NB (1997). Malonl-CoA regulation in skeletal muscle: its link to cell citrate and the glucose-fatty acid cycle. Am J Physiol 272: F641-8
104. Saltin B. Astrand P-O. Free fatty acids and exercise. Am J Clin Nutr 1993: 57: 7525-8 Saltin B. Gollnick PD (1983). Skeletal muscle adaptability: significance for metabolism and performance. Handbook of physiology. Baltimore. Williams and Wilkins. 555-661
105. Sidossis I.S. Gastaldelli A. Klain S. Wolfe RR (1996). Regulation of plasma fatty acid oxidation during low-and high-intensity exercise. Am J Physiol 272: E1065-70
106. Sidossis LS. Wolfre RR (1996). Glucose and insulin-induced inhibition of fatty acid oxidation: the glucose-fatty acid cycle reversed. Am J Physiol 270: E733-8
107. Spriet LI. Heigenhauser GJF, Jones NI (1986). Endogenous triacylglycerol utilization by rat skeletal muscle during titanic stimulation. J Appl Physiol 60: 410-5
108. Torres SH. Wikinski R. Dominguez J (1990). Effect of lipid infusion on substrate uptake in soleus muscle of the cat. Fisiología Acta Cient Ven 41: 33-9
109. Turcotte LP. Kiens B. Richter EA (1991). Saturation kinetics of palmitate uptake in perfused skeletal muscle. FEBS 279: 327-9
110. Turcotte LP. Richter EA. Kiens B (1992). Increased plasma FFA uptake and oxidation during prolonged exercise in trained vs, untrained humans. Am J Physiol 262: E791-9
111. Van Breda F. Keizer HA. Vork MM, Surtel DAM, de Jong YF, van der Vusse GF, Glatz JFC (1992). Modulation of fatty-acid-binding protein contento f Herat and skeletal muscle by endurance training and testoterone treatment. Pfluglers Arch 421: 274-9
112. Van der Vusse GJ. Reneman RS (1996). Lipid metabolism in muscle. Handbook oh Physiology section 12: Exercise: Regulation and integration of multiple systems. New York. Oxford Press. 952-94
113. Viru A (1992). Plasma hormones and physical exercise: a review. Int J Sports Med 13: 221-9
114. Vukovich MD. Costill DL. Hickey MS. Trappe SW. Cole KJ. Fink WJ (1993). Effect of fat emulsion infusion and fat freeding on muscle glycogen utilization during cycle exercise. J Appl Physiol 75: 1513-8
115. Wagenmakers AJM. A (1996). Malonyl-CoA fuel sensing mechanism in muscle: effects of insulin glucose and denervation. Commentary. Am J Physiol 269: E283-9
116. Walker M. Fulcher GR. Sum CF. Orskow H. Alberti GMM (1991). Effect of glycemia and nonesterified fatty acids on forearm muscle glucose uptake. Am J Physiol 261: E301-14
117. Winder WW. Arogyasami J. Elayan IM,. Vehrs PR (1989). Muscle malonyl-CoA decreases during exercise. J Appl Physiol 67: 2230-3
118. Winder WW. Arogyasami J. Elayan IM Cartmill D (1990). Time course of exercise-induced decline in malonyl-CoA in different muscle types. Am J Physiol 259: E266-71
119. Winder WW. Baldwin KM. Holloszy JO (1974). Enzymes involved in ketone utilization in different types of muscle: adaptations to exercise. Eut J Biochem 47: 461-7
120. Winder WW. Hickson RC. Hagberg JM. Ehsani JM. McLane JA (1979). Training induced changes in hormonal and metabolic responses to submaximal exercise. J Appl Physiol 46: 766-71
121. Wolfe BM. Klein S. Peters EJ Schmidt BF. Wolfe RR (1988). Effect of elevated free fatty acids on glucose oxidation in normal humans. Metabolism 37: 323-9
122. Yki-Jarvinen H. Puhakainen I. Koivisto VA (1991). Effect of free fatty acids on glucosa uptake and nonoxidative glicólisis across human forearm tisúes in the basal state and during insulin stimulation. J Clin Endocr Metab 72: 1268-77
123. Zorzano A. Balon TW, Brady Lj. Rivera P. Garetto LP. Young JC, Goodman MN, Ruderman NB (1985). Effects of starvation and exercise on concentrations of citrate, hexose phosphates and glycogen in skeletal muscle and heart. Bioch J 232: 285-91