
Una Perspectiva “Láctica” sobre el Metabolismo
Department of Kinesiology, Auburn University, Auburn, AL
Artículo publicado en el journal PubliCE del año 2014.
Publicado 28 de noviembre de 2014
Resumen
Palabras clave: Lactato, lanzadera, acidosis láctica, equilibrio acido base
INTRODUCCION
El estudio del metabolismo del lactato tiene sus orígenes en 1700 y, como en la mayoría de los campos de estudio en los últimos años, han surgido diferentes hipótesis. El propósito de esta breve revisión es presentar un resumen conciso de tres temas que están en la vanguardia de la investigación actual sobre el metabolismo del lactato: 1) el punto de vista innovador actual de la lanzadera de lactato célula a célula correctamente establecido en la actualidad, 2) las perspectivas antagónicas de la discutida lanzadera de lactato intracelular y 3) las diferentes opiniones sobre el papel del lactato (La-) en la acidosis que se produce durante el ejercicio. El tercer tema puede parecer algo dispar de los dos pero, al igual que la lanzadera de lactato intracelular, está siendo muy debatido en la actualidad y tiene amplias implicaciones.
LANZADERA CÉLULA A CÉLULA DE LACTATO
Lo que en la actualidad se conoce como lanzadera de lactato célula a célula fue presentado por Brooks (9) simplemente como lanzadera de lactato. Desde su introducción en 1984, esta hipótesis ha sido validada repetidamente por estudios que han utilizado una amplia variedad de metodologías experimentales. Postula que la formación de La- y su posterior distribución a lo largo del cuerpo es un mecanismo fundamental mediante el cual se puede llevar a cabo la coordinación del metabolismo intermediario en diferentes tejidos y células dentro de esos tejidos. La importancia del La- como fuente de combustible proveniente de carbohidratos se destaca por el hecho que durante el ejercicio de intensidad moderada, el flujo de La- sanguíneo puede superar al flujo de glucosa (71).
Debido a su gran masa y capacidad metabólica, el músculo de esquelético es probablemente el componente de mayor importancia para la lanzadera del lactato, no sólo en lo que se refiere a la producción de La- si no que también en lo que respecta al consumo neto y al uso de La-. La Figura 1 ilustra los resultados de un prolijo estudio realizado por Pagliassotti y Donovan (50). En este estudio realizado con músculos aislados de conejo perfundidos con una solución fisiológica que contenía glóbulos rojos bovinos, los autores midieron el balance neto de La- a lo largo de los músculos en diferentes niveles de concentraciones de La- ([La-]) perfundido. En reposo, tanto los músculos glucolíticos rápidos como los músculos oxidativos lentos liberaron La- en una cantidad neta, pero a medida que aumentaba la [La-] en el perfundido ambos tipos de músculo revirtieron a un consumo neto de La- -. Sin embargo, observaron que el músculo oxidativo revirtió a un consumo neto en una concentración del perfundido mas baja (~2.5 mM) en comparación con el músculo glucolítico (~4 mM).
En un estudio de mi propio laboratorio (29), nosotros observamos que el grupo muscular oxidativo gastrocnemius-plantaris-superficialis de perro (GP) in situ presentó un consumo neto de La- (~9 mM) cuando fue perfundido con una elevada [La-]. Adicionalmente, cuando la tasa metabólica aumentaba estimulando al músculo para que se contrajera, la tasa de consumo neto de La- aumentó junto con el consumo de oxígeno en estado estable
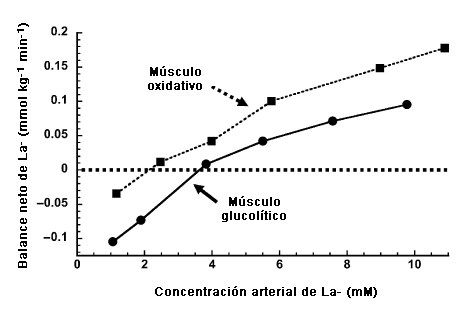
Figura 1. Balance neto del La- en músculos del conejo con diferentes tipos de fibra: glucolíticas (gracilis, ~99% de fibras de tipo IIb) y oxidativas (soleus, -98% tipo de fibras tipo I). Los valores por debajo de la línea punteada en 0 indican la liberación neta de La- mientras que los valores por encima de la línea indican el consumo neto de La-. Notemos que el musculo oxidativo cambia a un consumo neto en una menor [La-] perfundido (~2,5 mM) en comparación con el musculo glucolítico (~4 mM). Extraído y modificado con autorización de Pagliassotti y Donovan (50).
De manera similar, cuando aumenta la [La-] en sangre a valores de 2,5-4,0 mM o mayores, tanto los músculos que se encuentra realizando ejercicio como los que se encuentran en reposo se transforman en sitios de consumo neto.
En seres humanos, Richter et al. (55) caracterizaron una arteria y una vena femorales, midieron el flujo de sangre venosa femoral, y de evaluaron así el intercambio de La- en el músculo cuádriceps durante diferentes condiciones de ejercicio. Los cuádriceps liberaron La- durante el reposo y durante las contracciones. Sin embargo, cuando se agregaron ejercicios para los brazos a los ejercicios para las piernas, aumentó la concentración arterial de La- y los cuádriceps pasaron de producción neta a consumo neto de La- . Más recientemente, van Hall et al. (74) midieron el balance neto de La- en brazos y piernas de esquiadores de fondo de élite durante 40 min de esquí sobre ruedas (roller skiing) continuo. Durante este período de ejercicios para brazos y piernas, los brazos liberaron La-, mientras que las piernas incorporaron La-; el consumo neto de La- de la pierna se correlacionó estrechamente con la liberación de lactato. De manera similar, los estudios realizados en personas que están realizando ejercicios coinciden con los estudios efectuados con músculos animales aislados en los cuales, dependiendo de la concentración de La- en la sangre y de la tasa metabólica, se observó que los músculos pueden ser sitios primarios de consumo de La-. Además, como ha señalado Brooks (10), los músculos que se contraen in vivo o in situ típicamente liberan La- temporariamente después de que comienzan las contracciones y luego revierten ya sea a un intercambio neto de lactato igual a cero o a un consumo neto de La- a medida que las contracciones continúan. Este modelo ha sido denominado "efecto Stainsby" (10), en reconocimiento al hecho que el laboratorio de Wendell Stainsby fue uno de los primeros en informar este fenómeno (76).
¿Cuál es el destino del La- incorporado por el músculo? Nuevamente, utilizando el GP canino in situ, mi laboratorio (37) efectuó un seguimiento del destino metabólico del lactato [U-14C] tanto en reposo como durante las contracciones, creando una hoja de equilibrio. Tal como se presenta en la Figura 2, la mayoría de la marca (aprox. 55%) se mantuvo en el extracto muscular del músculo en reposo, quizás en forma de La-, piruvato, aminoácidos, o metabolitos en las vías metabólicas glucolíticas-gluconeogénicas y/o en las vías metabólicas de los ácidos grasos sin triacilglicerol (13). En los músculos en reposo, el resto de la marca de 14C apareció en el glucógeno, los aminoácidos y el piruvato liberados por el músculo, y en forma de 14CO2 (que representa la oxidación de La-) (37). En conjunto, parecería que el músculo en reposo solo podría oxidar La- lentamente debido a una tasa metabólica baja, y podría actuar como un depósito pasivo. En el músculo en reposo, la conversión neta de La- en glucógeno es más importante en los músculos que tienen un predominio de fibras de tipo glucolítico (18, 50). En el músculo que se está contrayendo, la historia es marcadamente diferente. Tal como se observa en la Figura 2, la gran mayoría (aprox. 83%) de la marca de 14C proveniente del La- aparece en forma de 14CO2, lo que representa la oxidación del La- en el músculo activo. Así, los músculos oxidativos que realizan ejercicios en intensidades moderadas en estado estable son ávidos consumidores de La- como combustible.
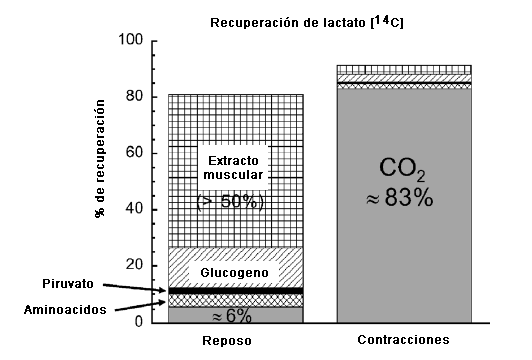
Figura 2. Destino del La- en un músculo gastrocnemio canino perfundido determinado a partir de la recuperación de lactato marcado radioactivamente con 14C ([14C]lactato). Nota 1) gran cantidad de oxidación de La- en el músculo contraído evidenciada por la recuperación de la marca 14C en CO2, 2) gran cantidad de marca radioactiva en el extracto muscular de musculo en reposo y 3) aparición significativa de marca radioactiva en el glucógeno de músculos en reposo. Basado en los datos de Kelley et al. (37).
Recientemente, estos resultados fueron confirmados y ampliados por estudios realizados en humanos mediante la técnica de clamp de lactato (infusión exógena de lactato). Usando una combinación de marcadores [3-13C]lactato, H13CO3-, y [6,6-2H2]glucosa, Miller et al (47,48) realizaron una investigación con sujetos que realizaban ejercicios a una intensidad de ejercicio moderada (aprox. 55% VO2max) con infusión de La- para mantener la concentración de lactato plasmática arterial venosa en aproximadamente 4 mM. De manera general, los autores (47) observaron un aumento significativo en la oxidación de La- acompañado por una disminución en la oxidación de glucosa; la interpretación que realizaron los autores fue que el La- compite exitosamente con la glucosa como fuente de combustible de carbohidratos, ahorrando así glucosa sanguínea para que pueda ser utilizada por otros tejidos como el cerebro. Adicionalmente, Miller et al. (48) observaron que aunque el La- reemplazaba hasta cierto punto a la glucosa como sustrato de la gluconeogénesis, la tasa absoluta de gluconeogénesis no se alteró por el clamp de lactato. En contraste, en un estudio de Roef et al. (60), el clamp de lactato aumentó la tasa gluconeogénica absoluta durante ejercicios de baja intensidad (~34% VO2max). En conjunto estos estudios demuestran que el La- se comportaba como un importante precursor en la gluconeogénesis tanto en ejercicios de intensidad baja como en ejercicios de intensidad moderada. Estos estudios con clamp, junto con muchas otras investigaciones de diferentes tipos, enfatizan el rol del La- como el sustrato sin duda más importante para la gluconeogenesis.
Los estudios sobre el balance de La- en el cuerpo entero sugieren que la remoción por parte del hígado explica aproximadamente 30% de la remoción total en seres humanos en reposo (14). Por medio de mediciones arterio-venosas en el hígado, Nielsen et al. (49) midieron un consumo hepato esplénico neto de La- de 0,4 mmol min-1 en sujetos en reposo. Durante el ejercicio realizado a aproximadamente 75% del VO2max, el flujo de sangre hepato-esplénico disminuyó de aproximadamente 1,6 a 0,7 L min-1 , pero el consumo neto de La- pasó a ser más del doble y alcanzó 1,0 mmol min-1 . Sin embargo, en los casos en donde el flujo de sangre disminuyó más notablemente, y la saturación de la oxihemoglobina hepato esplénica disminuyó a 6-10%, la diferencia arterio-venosa de La- se acercó a cero, lo que indica que la entrega de La- puede volverse limitante para la remoción del La- del hígado.
Debido a que el músculo cardíaco es mas oxidativo que incluso el músculo esquelético más oxidativo (34), no es sorprendente que el corazón sea un activo consumidor de La-. La evidencia proveniente de diferentes metodologías experimentales sugiere que a medida que aumentan la concentración de lactato sanguíneo, el flujo de sangre del miocardio y el VO2 del miocardio, el La- se transforma en el combustible preferido para el corazón, llegando a representar hasta un 60% del sustrato utilizado por el miocardio humano (69,23). Los estudios realizados con marcadores indican que esencialmente todo el La- incorporado por el corazón se oxida como combustible aeróbico (69).
Hasta el cerebro puede obtener La- de la sangre (17). La Figura 3 presenta la diferencia global en la concentración de La- arterio-venosa cerebral durante el descanso, el ejercicio incremental progresivo y la recuperación. A medida que la concentración arterial de La- aumenta progresivamente desde los valores de reposo (~1 mm) hasta aproximadamente 12 mM al final del ejercicio, y hasta aproximadamente 14 mM a comienzos de la recuperación antes de disminuir nuevamente hacia los valores observados en reposo, la diferencia arterio-venosa en el cerebro presenta esencialmente el mismo modelo. Aunque la contribución del consumo del cerebro con el consumo de La- por el cuerpo entero es despreciable, es de gran interés para la consideración de metabolismo del cerebro per se tal como se postula en la lanzadera de lactato de las neuronas tipo astrocito (51). Recientemente, Schurr (64) planteó la hipótesis que el La- es el producto principal de la glucolisis cerebral, ya sea que el cerebro se encuentre en reposo o en actividad y que las condiciones metabólicas sean aeróbicas o anaeróbicas. Como resultado, el La- se transforma en el combustible predominante para el ciclo de los ácidos tricarboxílicos (a través de la reconversión a piruvato), situación muy similar a lo que ocurre en el músculo esquelético.
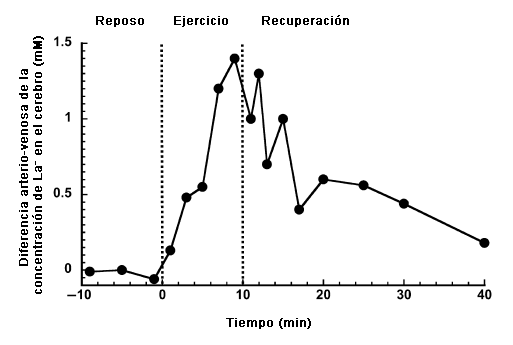
Figura 3. Diferencia global cerebral arterio-venosa de la concentración de La- durante el reposo, ejercicio incremental progresivo y la recuperación. Obtenido y modificado con permiso de Dalsgaard et al. (17).
Mediante estudios con marcadores isotópicos además de otras metodologías, arribamos al siguiente escenario. Es probable que las fibras musculares glucolíticas estén produciendo y liberando La- durante el ejercicio de intensidad moderada a alta. Mientras que algo de este La- pasa a la circulación, otra parte podría difundir a las fibras de los músculos oxidativos vecinos que pueden removerlo de la sangre y oxidarlo (8, 70). El La- liberado a la sangre puede ser tomado por las fibras de otros músculo esqueléticos que pueden estar en reposo o realizando ejercicio moderado. La mayoría del La- incorporado por los músculos esqueléticos es removido para ser utilizado como combustible oxidativo, y la tasa absoluta depende de la tasa metabólica de los músculos que se encuentran en reposo o realizando ejercicio (2, 37, 45, 70). Una parte del La- de la sangre es utilizado como combustible por el corazón, y otra parte probablemente se utiliza en el hígado durante la gluconeogenesis. Claramente, el intercambio de La- es un proceso dinámico donde se produce consumo y liberación muscular simultáneos durante el reposo y durante el ejercicio (9, 36, 70).
Luego de su introducción a principios de 1980, la lanzadera de lactato (célula a célula) se llamo "hipótesis" de la lanzadera de lactato. El concepto se ha extendido debido a la creciente evidencia de lanzaderas para neuronas astrocitos, lactato-alanina, peroxisomas, y lanzaderas de lactato espermatogénicas que pueden ser consideradas subcomponentes de la lanzadera de lactato célula a célula general (Consultar Gladden (26) para obtener referencias). En este modelo, el La- es un intermediario importante en los numerosos procesos metabólicos, un combustible móvil para el metabolismo aeróbico, y quizás un mediador de estado redox entre los diferentes compartimientos dentro de las células y entre las células. En la Figura 4 se ilustran los aspectos esenciales de la lanzadera de lactato célula a célula. Para obtener detalles adicionales consultar la revisión de Gladden (26). Debido al hecho que el apoyo experimental para la lanzadera de lactato es esencialmente unánime, es tiempo de quitar la denominación de "hipótesis." La lanzadera de lactato célula a célula en la actualidad está establecida como el mapa del metabolismo del La- para todo el cuerpo.
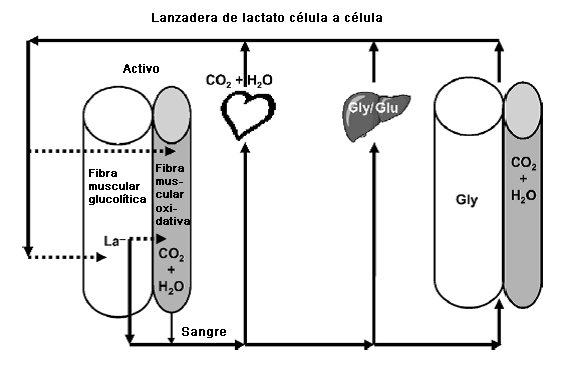
Figura 4. Esquema de la lanzadera de lactato célula a célula. Los cilindros de color claro representan fibras musculares glucolíticas, y los cilindros más oscuros representan fibras musculares oxidativas. Las fibras en la izquierda del diagrama representan los músculos activos, mientras que las fibras de la derecha representan el músculo en reposo. Las fibras glucolíticas producen La- más rápidamente. La flecha discontinua de la fibra glucolítica activa hacia la fibra oxidativa activa indica la posibilidad de movimiento del La- desde una fibra productora de La- hacia una fibra consumidora de La-. Las flechas sólidas indican que el flujo sanguíneo distribuye el La- en todo el cuerpo; por ejemplo, al corazón para que sea oxidado como combustible, al hígado para que sea almacenado como glucógeno, o para que sea convertido en glucosa y sea liberada (ciclo de Cori). Las fibras del músculo inactivas ubicadas a la derecha pueden incorporar La- para la posible síntesis de glucógeno en las fibras glucolíticas y para que en menor grado sea oxidado en las fibras oxidativas. Las flechas discontinuas de la izquierda indican que el La- puede volver hacia los músculos activos. La pequeña flecha sólida de la fibra de músculo oxidativo activo indica que con ejercicio intenso, estas fibras también pueden producir y liberar La-. Para obtener detalles adicionales consultar el texto.
LANZADERA INTRACELULAR DE LACTATO
En 1998 Brooks propuso la existencia de una lanzadera de lactato intracelular (10) y aportó datos que corroboraban esto en un trabajo de investigación de 1999 (12). Un principio central de esta lanzadera intracelular fue que el La- es un producto inevitable de la glucolisis, particularmente durante la glucolisis rápida, porque la lactato deshidrogenasa (LDH) tiene una Vmax más alta que cualquier enzima en la vía glucolítica, y porque la Keq para la conversión de piruvato a La- se encuentra bastante desplazada en dirección al La- y NAD+ (8,10-12). Con esta información, Brooks et al. (11) cuestionaron cómo sería posible que el La- se convirtiera nuevamente a piruvato en el citosol, permitiendo así que los tejidos bien perfundidos oxidaran el La-. El grupo de Brooks (11, 12, 19) ha aportado evidencia de los siguientes componentes fundamentales de una lanzadera de lactato intracelular en el músculo esquelético: 1) consumo y oxidación directos de La- por parte de mitocondrias aisladas sin conversión previa extra mitocondrial de La- a piruvato, 2) presencia de un pool de LDH intra mitocondrial, y 3) presencia del transportador de La- llamado transportador 1 monocarboxilato (MCT1) en la mitocondria, probablemente en la membrana mitocondrial interna. El caso completo fue presentado por Hashimoto y Brooks (30) en otro trabajo de investigación de este simposio.
El funcionamiento de una lanzadera de lactato intracelular como la descripta por el grupo de Brooks (26, 28) vincula la producción constante de La- en el citosol con el incremento en la tasa de producción con la actividad glucolítica elevada. Debido a su elevada concentración en relación al piruvato, el La- sería el principal monocarboxilato en difundir hacia la mitocondria. En la versión original de esta lanzadera (Figura 5 en la referencia (12)), este La- seria transportado luego a través de la membrana mitocondrial interna por MCT1. Una vez dentro de la matriz mitocondrial, la LDH mitocondrial catalizaría la conversión de La- a piruvato que se oxidaría a acetil CoA a través de la reacción PDH. Luego el acetil CoA ingresaría al ciclo de los ácidos tricarboxílicos (TCA). Un punto importante es que esta lanzadera de lactato intracelular no sólo entregaría el sustrato en forma de La- para su conversión a piruvato; si no que también entregaría los equivalentes reducidos (NADH), suplantando en diferentes grados el papel de las lanzaderas malato-aspartato y glicerol-fosfato, dependiendo de la tasa de formación de La- y su velocidad de transporte hacia adentro de la mitocondria.
¿Las mitocondrias pueden oxidar directamente el La-? Aunque Brooks et al (12) aportaron datos más completos en 1999 y plantearon la hipótesis, Szczesna-Kaczmarek (73) fue realmente el primero en informar la oxidación mitocondrial directa del La- en el músculo esquelético, en 1990. Sin embargo, poco después de esto, Popinigis et al. (53) observaron oxidación no acoplada de La- en el músculo esquelético humano. Adicionalmente, desde que Brooks propuso por primera vez la lanzadera de lactato intracelular, ha surgido evidencia en contra de la misma. Rasmussen et al. (54) y Sahlin et al. (62) no observaron ninguna evidencia que las mitocondrias puedan usar La- como sustrato sin la conversión previa a piruvato en el citosol. Un informe preliminar realizado por Willis et al. (77) también reportó una actividad insignificante de la lanzadera de lactato intracelular propuesta en mitocondrias aisladas de músculo esquelético (fibras de tipo I y IIb) y en hígado de ratas. Finalmente, Yoshida et al. (81) del laboratorio de Bonen también observaron recientemente una oxidación directa mínima de La- por las mitocondrias del subsarcolema o de las fibras musculares del músculo esquelético rojo (oxidativo) o blanco (glucolítico). Los méritos y las conclusiones del trabajo publicado por Yoshida et al. (81) han sido debatidos extensivamente en cartas al editor (3,7) y en una columna de Perspectivas (25) en el Journal of Physiology.
¿La LDH está presente dentro de las mitocondrias? Usando una combinación de técnicas histoquímicas y de microscopía electrónica, Baba y Sharma (1) localizaron la LDH en las mitocondrias de corazón y músculo esquelético de ratas. Posteriormente, Kline et al. (42) y Brandt et al. (6) utilizaron técnicas de fraccionamiento celular para demostrar la presencia de LDH en las mitocondrias de hígado, riñón y corazón de ratas. Brooks citó evidencia de su propio trabajo (12, 19) y estos estudios previos como respaldo de la presencia de LDH en las mitocondrias, y argumentó que otros trabajos (Rasmussen et al. (54) y Sahlin et al. (62)) probablemente perdieron la LDH en los procedimientos de aislamiento para obtener las mitocondrias. No es sorprendente que, Rasmussen et al. (54) y Sahlin et al. (62) utilizaran los estudios anteriores para apoyar su posición y expresaran que las mitocondrias de Brooks et al. (12,19) se contaminaron con LDH citosólica. Nuevamente, Yoshida et al. (81) presentaron recientemente evidencia contra la presencia de LDH dentro de las mitocondrias tanto del subsarcolema como intermiofibrilares del músculo oxidativo y del musculo glucolítico.
En mi opinión, el peso de la evidencia está en contra de la presencia de LDH en la matriz mitocondrial y de la oxidación de La- dentro de la matriz mitocondrial. Sin embargo, esto no significa que no haya una lanzadera de lactato intracelular, ni excluye la posibilidad que la LDH mitocondrial se localice en el espacio intermembranoso (una posibilidad ya considerada por Brandt y Kline y colegas en 1986-1987 (6, 42)), quizás se encuentre acoplada al exterior de la membrana mitocondrial interna (31). De hecho, sería bastante posible, incluso probable que funcione una lanzadera como esta, aunque sin metabolismo de lactato dentro de la matriz. Es razonable especular que las concentraciones de piruvato y de NADH sean menores en cercanías de las mitocondrias donde el transportador de piruvato (un MCT?) y la lanzadera de NADH (malato-aspartato y gicerol fosfato) transportan el piruvato y los equivalente de NADH, respectivamente, hacia adentro de la mitocondria. En otros términos, las mitocondrias que realizan una oxidación activa crearían sumideros para el uso de piruvato y NADH y, por consiguiente, su captación a partir de ubicaciones citosólicas adyacentes. Al mismo tiempo, los sitios de glucolisis celular crearían concentraciones crecientes de La-; el principal producto final de la glucolisis sería el La- producido por la gran actividad de la LDH, tal como se describió previamente. Esta situación produciría una mayor producción y concentración de La- en los compartimientos citosólicos alejados de las mitocondrias. Entonces, debido a la relativamente superior [La-] en comparación con la [piruvato], el La- sería la principal especie en difundir a las áreas cercanas a las mitocondrias. La concentración de La- típicamente es aproximadamente 10-200 veces mayor que la concentración de piruvato en las biopsias de músculo esquelético (63). Al lado de las mitocondrias, el La- y NAD+ serían convertidos nuevamente a piruvato y NADH por la LDH para ser incorporados a las mitocondrias. Un esquema como este permitiría acoplar la producción de La- con la posterior oxidación y permitiría un menor transporte de La- hacia afuera de la célula. Esta hipótesis alternativa de lactato intracelular fue originalmente planteada por Stainsby y Brooks (68) en 1990. También debemos destacar que Yoshida et al. (81) informaron que la [La-] tendría que ser de un orden 69-139 veces mayor que la concentración de piruvato para que mitocondrias aisladas alcancen una tasa de oxidación similar, una proporción que puede ocurrir en el músculo esquelético durante la recuperación frente al ejercicio intenso (63).
El modelo descrito arriba es consistente con la compartimentalización del metabolismo tal como se describió en varios estudios. James y colegas (33) propusieron que la bomba Na+/K+-ATPasa obtiene su energía mayormente de la glucolisis que está estrechamente asociada con la bomba; esta idea ha sido apoyada recientemente en los estudios realizados con fibras del músculo esquelético desolladas mecánicamente (20). Varios investigadores han presentado evidencia que también existe una compartimentalización funcional de la glucolisis con el retículo sarcoplásmico (21). ¿Es posible que el La- proveniente de la glucolisis asociado con la actividad de la Na+/K+-ATPasa se desvíe hacia la mitocondria del subsarcolema, mientras que el La- proveniente de la glucolisis asociada con el bombeo sarcoplásmico de Ca2+ sea desviado hacia las mitocondrias intermiofibrilares? Quizás los estudios futuros nos aporten evidencia al respecto.
Observaciones recientes de Hashimoto et al. (31) del laboratorio de Brooks aportaron otra base estructural del funcionamiento de una lanzadera de lactato intracelular fuera de la matriz. Empleando técnicas de microscopia confocal láser, y de inmunotransferencia después de inmunoprecipitación en células L6 de músculo esquelético, Hashimoto et al. (31) observaron evidencia que sugiere que la LDH, los MCT1, las glucoproteínas transmembranales de un solo paso CD 147 y la citocromo oxidasa se localizarían conjuntamente en la membrana mitocondrial interna, y que la enzima LDH aparentemente se ubicaría en la superficie exterior de la membrana interna. Los autores (31) llamaron a esta asociación, complejo de oxidación del lactato. Hashimoto y Brooks (30) enumeraron la evidencia experimental que respalda la existencia de este complejo en otro trabajo presentado en este simposio, y lo ilustran en la Figura 1. Que un complejo de este tipo pueda facilitar el funcionamiento de una lanzadera de lactato intracelular, no es una condición excluyente para el proceso. No obstante, la ubicación potencial de la LDH levemente adherida al exterior de la membrana mitocondrial interna vuelve a encender la posibilidad de que tal LDH podría ser susceptible a la pérdida durante el aislamiento de las mitocondria del tejido muscular. Probablemente, los estudios futuros nos aportarán evidencia conclusiva con respecto a la pérdida de LDH versus contaminación en experimentos realizados con mitocondrias aisladas.
ACIDOSIS LÁCTICA
Uno de los primeros acercamientos fisicoquímicos del equilibrio acido base en sangre humana tenía la forma de la ecuación de Henderson-Hasselbalch (1918, citado por Corey (16)):
pH = pK' + log [HCO3-] / (SCO2 x PCO2)
donde pH es el logaritmo negativo de [H+], pK es el logaritmo negativo de la constante de disociación de ácido carbónico a bicarbonato e ion hidrógeno, SCO2 es el coeficiente de solubilidad del dióxido de carbono (CO2) en el plasma, y PCO2 es la presión parcial de CO2 en el plasma. En este ejemplo de equilibrio ácido-base, la [H+] en el plasma está regulada/determinada por dos variables, PCO2 y [HCO3-]. A finales del siglo 20, Siggaard-Andersen (65) reconoció que la ecuación de Henderson-Hasselbalch por si sola aporta una descripción insuficiente del equilibrio ácido-base y estableció el concepto de exceso de bases (16). El exceso de bases es el número de miliequivalentes de ácido o base que se necesitan para valorar 1 L de sangre hasta un pH 7,40 a 37°C y a una PCO2 de 40 mmHg. Posteriormente, el exceso de base se volvió el enfoque predominante al equilibrio ácido-base clínico (16, 78). Tal como lo señaló Corey (16), el enfoque del exceso de base describe el equilibrio ácido-base en base a dos variables, el exceso de base y la PCO2, y tiene varios rasgos esenciales: 1) la cantidad de H+ agregados o removidos de la sangre u otros fluidos se considera que determina la [H+] final y 2) las membranas plasmáticas pueden ser permeables a H+ (o HCO3-), lo que permite que las reacciones químicas intracelulares y extracelulares influencien la [H+].
En el contexto de producción y consumo de H+, se ha establecido un concepto erróneo (27) en el campo de la fisiología del ejercicio y, quizás también en otras áreas de estudio. Esta falacia es que la especie involucrada en las reacciones bioquímicas es el ácido láctico (HLa). En otros términos, es el acido láctico quien se produce o se consume bioquímicamente en la reacción de la LDH. En consecuencia, el error se transfirió hacia el equilibrio ácido-base de la siguiente manera. Dado que el HLa se produce como producto final de la glucolisis, sobre todo durante el ejercicio intenso, y el 99.5% del HLa se encuentra disociado en los valores de pH fisiológicos, la explicación fue que se producía HLa y luego se disociaba para liberar H+, lo que disminuía el pH y provocaba una acidosis láctica intracelular.
Recientemente, Robergs et al. (57-59) detallaron cuidadosamente las reacciones de la glucolisis, incluyendo el papel del H+ en cada paso, y establecieron que la reacción catalizada por la LDH produce La-, no HLa; en esta conclusión, ellos están en lo cierto. En una revisión de ideas publicadas previamente (32), Robergs et al. (57-59) enfatizaron el punto de vista que la producción de La- en la reacción de LDH consume H+ y, en su opinión, realmente mitiga la acidosis intracelular. No nos sorprende que esto provocara un gran debate (4, 40, 41, 44).
En el centro de esta controversia está la pregunta de causa y efecto. ¿Cuáles son las variables independientes y cuales las variables dependientes en el equilibrio ácido-base?. Parece haber acuerdo con la idea que el PCO2 (o, en algunos casos, contenido total de CO2) es uno del determinantes independientes de [H+] (16, 38, 66). Desde el punto de vista de Siggaard-Andersen (65,66) (como mencionamos anteriormente) y Robergs et al. (57), la producción/adición o consumo/remoción de protones también es un factor de control para la determinación de la [H+] final. Los científicos que estudian las lanzaderas subcelulares también se focalizan en la transferencia de H+ (y HCO3-) por las membranas como agente causal (5). Sin embargo, Stewart (72) aportó una visión fisicoquímica alternativa que tenía tres variables determinantes independientes: 1) PCO2 (o contenido total de CO2 en un sistema cerrado), 2) concentración total de ácidos débiles ([Atot]), y 3) diferencia de iones fuertes ([SID]). El PCO2 fue definido antes. [Atot] es la concentración total de electrolitos/ácidos débiles en un fluido corporal dado. [SID] es la diferencia entre la suma de las concentraciones de todos los cationes de bases fuertes y la suma de las concentraciones todos los aniones ácidos fuertes (por ejemplo, [Na+] + [K+] + [Ca2+] - [Cl-] - [La-]). Debido a problemas con el espacio, el modelo de Stewart no será presentado con detalle en esta publicación; el lector interesado puede consultar otras fuentes (22, 35, 44, 72).
En la postura de Robergs sobre la acidosis (57), los protones se producen y consumen en las diferentes reacciones de la glucolisis; el ATP se forma en las reacciones no mitocondriales y es hidrolizado para mantener la energía para las contracciones/ejercicio y el La- se acumula a través de la reacción de la LDH. En este paradigma, la acidosis es el resultado de la hidrolisis no mitocondrial del ATP que produce protones mientras que la formación de La- realmente limita la acidosis consumiendo los protones. Para Robergs (57), no existe ninguna condición como la acidosis láctica. Al contrario, en el modelo de Stewart (72), el La-es un ion fuerte, y su acumulación reduce la [SID], lo que provoca alteraciones en el equilibrio de disociación del agua y de los ácidos débiles; el resultado final es que la acumulación de La- es al menos parcialmente la causa de la acidosis, parte de la cual podría llamarse acidosis láctica. Además, Stewart (72) ha defendido enérgicamente y repetidamente que la [H+] no está determinada por la cantidad de H+ agregados o removidos de una solución, algo que postulan otros autores (22, 35, 44). "No importa cual sea la fuente bioquímica de protones, estas variables ácido-base dependientes no pueden ser consideradas como factores independientes para determinar sus propias concentraciones" (44).
El modelo Stewart ha planteado numerosos aspectos fascinantes, tales como si los iones H+ u OH- individuales realmente participan sustantivamente en reacciones bioquímicas o procesos de transporte de membrana (44). El modelo Stewart también sostiene que [SID] es el principal mecanismo por el cual los fluidos del cuerpo pueden afectar los valores de [H+] unos de otros (22, 72). Se han presentado datos que demuestran los cambios en la concentración de iones fuertes en el músculo que ha realizado ejercicio y la liberación y captación de iones hacia y desde el músculo que realizó ejercicio; se sostiene que los cambios resultantes en [SID] provocan cambios ácido-base en el músculo y en la sangre (35). Adicionalmente, el modelo Stewart ha ganado aceptación entre médicos que tratan a pacientes extremamente enfermos (38, 39).
Para finalizar, debemos destacar que el análisis de Stewart no era completamente nuevo. De manera significativa, Stewart introdujo nuevamente, clarificó e informatizó el concepto de análisis fisicoquímico del estatus acido base de los fluidos corporales, retomando los pensamientos de Henderson y Van Slyke y de otros investigadores del equilibrio ácido-base menos conocidos de principios del siglo 20 (35, 43). Es más, [SID] es la “base buffer" propuesta por Singer y Hastings en 1948 (66, 67, 78).
En el acalorado debate, a veces se genera más calor que luz. En ese contexto, es necesario tener presente que todos los enfoques sobre el equilibrio ácido-base se plantean a partir de la misma base común y dan las mismas respuestas en lo que se refiere a los parámetros ácido-base calculados (15, 79). Citando a Wooten (79): "Todos los modelos de equilibrio ácido-base utilizan el mismo concepto básico. Bajo el supuesto de equilibrio o en una aproximación al estado estable de equilibrio, alguna propiedad del sistema (por ejemplo, número de protones, sitios de unión a protones o cargas, entre otras posibles propiedades) se describe a partir de la distribución de esa propiedad entre las diferentes especies que comprenden el sistema, según la energética del sistema manifestada a través de las constantes de equilibrio relevantes de las diferentes especies bajo una serie dada de condiciones". Por ello, tanto el modelo tradicional (los sitios aceptadores de protones) como el modelo Stewart (carga eléctrica) pueden ser utilizados para calcular el pH en un estado contra otro (79), y cuando las medidas se toman en dos estados de equilibrio diferentes, los resultados son independientes del camino tomado entre las dos condiciones (16, 46, 79). Existe consenso con respecto a que el modelo de Stewart en particular al evaluar el equilibrio ácido-base, enfatiza la importancia de considerar todas las variables; por ejemplo, las seis ecuaciones de Stewart relacionadas a 1) equilibrio de la formación del ion bicarbonato, 2) equilibrio de la formación de carbonato, 3) equilibrio de disociación del agua, 4) balance de cargas eléctricas, 5) equilibrio de disociación del ácido débil, y 6) conservación de la masa (15, 72, 79). Otro método para contabilizar la [H+] es contar todas las liberaciones e incorporaciones de protones individuales en un sistema durante la transformación de un estado a otro (46, 75); éste es el método preferido por Robergs et al. (57).
Por lo tanto, nos queda la pregunta de que es lo que provoca una [H+] particular?. El enfoque de Stewart afirma que son las tres variables independientes: PCO2, [Atot] y [SID]. Robergs et al. (57) enfatizan la producción/adición y consumo/remoción de H+. Wooten (79, 80) afirman que 1) el caso de Stewart (y, por extrapolación, cualquier otro caso) para las variables independientes " carece de apoyo" experimental”; que 2) sin datos mecánicos reales, el debate de causalidad es “más filosófico que fisiológico"; y que 3) esta pregunta no puede ser contestada exclusivamente en base a la termodinámica. No obstante, Corey (16) sintetiza algunos ejemplos de evidencia que podrían apoyar la aserción del modelo de Stewart de que cambios en SID provocan cambios en la posición del equilibrio del agua y cambios en la [H+]. Como ejemplo, la carga iónica puede romper los puentes hidrógeno y afectar las propiedades del agua. En este sentido se necesitan estudios adicionales y estudios de nivel molecular en los cuales se analicen los transportadores de protones y de bicarbonato con gran detalle (56). Si bien se ha puesto énfasis en los protones y en el bicarbonato, un análisis mas detallado revela que los iones fuertes participan como especies en el transporte simporte o antiporte (56). Seguramente estudios mas detallados revelarán cuales iones realmente están siendo regulados y, por consiguiente, son de naturaleza causal.
Aproximadamente 80-100% de la transferencia de lactato por el sarcolema se produce ya sea por difusión de HLa no disociado o por transporte facilitado con MCT (24, 52, 61). En ambos casos, un protón es transferido con (o se une al) La-. ¿Cuál es la importancia de este proceso desde los músculos que realizan ejercicio hasta el plasma?. A partir de la perspectiva de Stewart, se incorpora al plasma un ion fuerte (La-) lo que causaría un aumento en la [H+]. A partir de la perspectiva de acido base tradicional de Robergs, se incorpora un protón al plasma lo que causaría un aumento en [H+]. Por consiguiente, ambos puntos de vista sugieren una acidosis láctica en el plasma.
Dado 1) el rol central de La- en la diferencia entre el estado de reposo y el estado de ejercicio tanto en músculo como en sangre, 2) la posibilidad que el La- pueda desempeñar un rol causal como ion fuerte, 3) la incertidumbre global sobre causa y efecto en el equilibrio ácido-base, y 4) la compatibilidad entre los diferentes modelos con respecto al efecto de HLa/La- en el equilibrio ácido-base del plasma, en este momento no hay ninguna razón convincente para cambiar la terminología de “acidosis láctica" por "acidosis hidrolítica de ATP no mitocondrial”.
CONCLUSIONES
En conclusión, esta breve revisión ha resumido el estado actual de tres tópicos correspondientes al estudio del metabolismo del La-: 1) la lanzadera de lactato célula a célula, 2) la lanzadera de lactato intracelular y 3) el papel del La- en la acidosis durante el ejercicio. Con respecto a la lanzadera de lactato célula a célula, existe un veredicto: El La- es un jugador central en el metabolismo celular, regional y de todo el cuerpo. Lejos de ser un metabolito sin salida, el La- es un punto de intersección/encrucijada en las vías del metabolismo. La lanzadera de lactato célula a célula ha sido repetidamente validada por numerosos estudios experimentales, y en la actualidad nos aporta el contexto para la interpretación del metabolismo global del lactato de todo el cuerpo.
En contraste, la lanzadera de lactato intracelular está siendo fuertemente debatida. Parecería probable que el La- es el producto principal de la glucolisis, particularmente en compartimientos intracelulares que están alejados de las mitocondrias. Probablemente la mayor parte de este La- difunde hacia las áreas adyacentes a las mitocondrias, quizás al espacio intermembranoso cerca del complejo de oxidación de lactato localizado dentro de la membrana mitocondrial interna. Es muy probable que la reconversión de La- a piruvato se produzca fuera de la membrana mitocondrial interna y no en la matriz mitocondrial. Los estudios futuros establecerán con mayor claridad el papel del La- intracelular en el metabolismo oxidativo del músculo esquelético.
Otra área polémica en el metabolismo del La- es el papel del La- en la acidosis del ejercicio. Durante gran parte del último siglo, la producción de ácido láctico y su posterior disociación han sido consideradas un factor importante en la acidosis por ejercicio intenso. Recientemente, una evaluación cuidadosa de las reacciones de la vía glucolítica introdujo nuevamente el concepto que es el La-, no el ácido láctico, el producto de la reacción catalizada por la LDH, y que la formación de La- realmente atenúa la acidosis. Esta visión no coincide con la noción que el La- es un contribuyente importante para la [SID] que es considerada por algunos un determinante independiente de [H+]. Una revisión de la literatura revela que los diferentes enfoques del equilibrio ácido base surgen de la misma base común y dan las mismas respuestas en lo que respecta a los parámetros ácido base calculados. Es sorprendente que no exista acuerdo general sobre las causas / los mecanismos reales de [H+]. No obstante en este momento, no hay evidencia suficiente para eliminar el término "acidosis láctica".
Referencias
1. Baba N. , Sharma H. M. (1971). Histochemistry of lactic dehydrogenase in heart and pectoralis muscles of rat. J. Cell. Biol. 51:621-35.
2. Bergman B. C. , Horning M. A. , Casazza G. A. , Wolfel E. E. , Butterfield G. E. , Brooks G. A. (2000). Endurance training increases gluconeogenesis during rest and exercise in men. Am. J. Physiol. Endocrinol. Metab. 278:E244-51.
3. Bonen A. , Hatta H. , Holloway G. , Spriet L. , Yoshida Y. (2007). Response to letter entitled "Investigation of the lactate shuttle in skeletal muscle mitochondria". J. Physiol. 584:707-8.
4. Boning D. , Strobel G. , Beneke R. , Maassen N. (2005). Lactic acid still remains the real cause of exercise-induced metabolic acidosis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289:R902-3.
5. Boron W. F. (2006). Acid-base transport by the renal proximal tubule. J. Am. Soc. Nephrol. 17:2368-82.
6. Brandt R. B. , Laux J. E. , Spainhour S. E. , Kline E. S. (1987). Lactate dehydrogenase in rat mitochondria. Arch. Biochem. Biophys. 259: 412-22.
7. Brooks G. , Hashimoto T. (2007). Investigation of the lactate shuttle in skeletal muscle mitochondria. J. Physiol. 584:705-6.
8. Brooks G. A. (2000). Intra and extra-cellular lactate shuttles. Med. Sci. Sports Exerc. 32(4):790-9.
9. Brooks G. A. (1985). Lactate: glycolytic product and oxidative substrate during sustained exercise in mammals—the 'lactate shuttle. ' In: Gilíes R, editor. Comparative Physiology and Biochemistry: Current Topics and Trends, vol. A, Respiration-Metabolism-Circulation. Berlin (Germany): Springer; p. 208-18.
10. Brooks G. A. (1998). Mammalian fuel utilization during sustained exercise. Comp. Biochem. Physiol. B. Biochem. Mol. Biol. 120:89-107.
11. Brooks G. A. , Brown M. A. , Butz C. E. , Sicurello J. P. , Dubouchaud H. (1999). Cardiac and skeletal muscle mitochondria have a monocarbox-ylate transporter MCT1. J. Appl. Physiol. 87:1713-8.
12. Brooks G. A, Dubouchaud H. , Brown M. , Sicurello J. P. , Butz C. E. (1999). Role of mitochondrial lactate dehydrogenase and lactate oxidation in the intracellular lactate shuttle. Proc. Nati. Acad. Sci. USA. 96:1129-34.
13. Chin E. R. , Lindinger M. I. , Heigenhauser G. J. (1991). Lactate metabolism in inactive skeletal muscle during lactacidosis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 261:R98-105.
14. Connor H. , Woods H. F. (1982). Quantitative aspects of L(+)-lactate metabolism in human beings. In: Porter R, Lawrenson G, editors. Metabolic Acidosis (Ciba Foundation Symposium 87). London (UK): Pitman Books Ltd. . p. 214-34.
15. Corey H. E. (2005). Bench-to-bedside review: fundamental principles of acid-base physiology. Crit. Care. 9:184-92.
16. Corey H. E. (2003). Stewart and beyond: new models of acid-base balance. Kidney Int. 64:777-87.
17. Dalsgaard M. K. , Quistorff B. , Danielsen E. R. , Selmer C. , Vogelsang T. , Secher N. H. (2004). A reduced cerebral metabolic ratio in exercise reflects metabolism and not accumulation of lactate within the human brain. J. Physiol. 554:571-8.
18. Donovan C. M. , Pagliassotti M. L. (2000). Quantitative assessment of pathways for lactate disposal in skeletal muscle fiber types. Med. Sci. Sports. Exerc. 32(4):772-7.
19. Dubouchaud H. , Butterfield G. E. , Wolfel E. E. , Bergman B. C. , Brooks G. A. Endurance training, expression, and physiology of LDH, MCL1, and MCL4 in human skeletal muscle. (2000). Am. J. Physiol. Endocrinol. Metab. 278:E571-9.
20. Dutka L. L. , Lamb G. D. (2007). Na+-K+ pumps in the transverse tubular system of skeletal muscle fibers preferentially use ATP from glycolysis. Am. J. Physiol. Cell. Physiol. 293:C967-77.
21. Entinan M. L. , Keslensky S. S. , Chu A. , Van Winkle W. B. (1980). The sarcoplasmic reticulum-glycogenolytic complex in mammalian fast twitch skeletal muscle. Proposed in vitro counterpart of the contraction-activated glycogenolytic pool. J. Biol. Chem. 255:6245-52.
22. Fencl V. , Leith D. E. (1993). Stewart's quantitative acid-base chemistry: applications in biology and medicine. Respir. Physiol. 91: 1-16.
23. Gertz E. W. , Wisneski J. A. , Stanley W. C. , Neese R. A. (1988). Myocardial substrate utilization during exercise in humans. Dual carbon-labeled carbohydrate isotope experiments. J. Clin. Invest. 82:2017-25.
24. Gladden L. B. (1996). Chapter 14. Lactate transport and metabolism during exercise. In: Rowell LB, Shepherd JT, editors. Exercise: Regulation and Integration of Multiple Systems. New York (NY): Oxford University Press;. p. 614-48.
25. Gladden L. B. (2007). Is there an intracellular lactate shuttle in skeletal muscle? J. Physiol. 582:899.
26. Gladden L. B. (2004). Lactate metabolism: a new paradigm for the third millennium. J. Physiol. 558:5-30.
27. Gladden L. B. (1989). Lactate uptake by skeletal muscle. Exerc. Sport. Sci. Rev. 17:115-55.
28. Gladden L. B. (2001). Lactic acid: new roles in a new millennium. Proc. Nati. Acad. Sci. USA. 98:395-7.
29. Gladden L. B. (1991). Net lactate uptake during progressive steady-level contractions in canine skeletal muscle. J. Appl. Physiol. 71: 514-20.
30. Hashimoto T, Brooks G. A. (2008). Mitochondrial lactate oxidation complex and an adaptive role for lactate production. Med. Sci. Sports. Exerc. 40(3):486-94.
31. Hashimoto T. , Hussien R. , Brooks G. A. (2006). Colocalization of MCT1, CD147, and LDH in mitochondrial inner membrane of L6 muscle cells: evidence of a mitochondrial lactate oxidation complex. Am. J. Physiol. Endocrinol. Metab. 290:E123 7-44.
32. Hochachka P. W. , Mommsen T. P. (1989). Protons and anaerobiosis. Science. 219:1391-7.
33. James J. H. , Luchette F. A. , McCarter F. D. , Fischer J. E. (1999). Lactate is an unreliable indicator of tissue hypoxia in injury or sepsis. Lancet. 354:505-8.
34. Jansson E. , Sylven C. (1986). Activities of key enzymes in the energy metabolism of human myocardial and skeletal muscle. Clin Physiol. 6:465-71.
35. Johnson R. L Jr. , Heigenhauser G. J. F. , Hsia C. C. W. , Jones N. L. , Wagner P. D. (1996). Determination of gas exchange and acid-base balance during exercise. In: Rowell LB, Shepherd JT, editors. Handbook of Physiology. Section 12: Exercise: Regulation and Integration of Multiple Systems. New York (NY): Oxford University Press. p. 515-84.
36. Jorfeldt L. (1970). Metabolism of L(+)-lactate in human skeletal muscle during exercise. Acta Physiol. Scand. Suppl. 338: 1-67.
37. Kelley K. M. , Hamann J. J. , Navarre C. , Gladden L. B. (2002). Lactate metabolism in resting and contracting canine skeletal muscle with elevated lactate concentration. J. Appl. Physiol. 93:865-72.
38. Kellum J. A. (2005). Clinical review: reunification of acid-base physiology. Crit. Care. 9:500-7.
39. Kellum J. A. (1998). Metabolic acidosis in the critically ill: lessons from physical chemistry. Kidney Int. Suppl. 66:S81-6.
40. Kemp G. (2004). Lactate accumulation, proton buffering, and pH change in ischemically exercising muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289:R895-901.
41. Kemp G. , Boning D. , Beneke R. , Maassen N. (2005). Explaining pH change in exercising muscle: lactic acid, proton consumption, and buffering vs. strong ion difference. Am. J. Physiol. Regul. Integr. Comp. Physiol. 291:R235-7.
42. Kline E. S. , Brandt R. B. , Laux J. E. , et al. (1986). Localization of L-lactate dehydrogenase in mitochondria. Arch. Biochem. Biophys. 246:673-80.
43. Lindinger M. I. (2003). Exercise: a paradigm for multi-system control of acid-base state. J Physiol. 550:334.
44. Lindinger M. I. , Kowalchuk J. M. , Heigenhauser G. J. (2005). Applying physicochemical principles to skeletal muscle acid-base status. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289:R891-4.
45. Mazzeo R. S. , Brooks G. A. , Schoeller D. A. , Budinger T. F. (1986). Disposal of blood [1- C]lactate in humans during rest and exercise. J. Appl. Physiol. 60:232-41.
46. Meyer R. A. , Wiseman R. W. (2006). The metabolic systems: control of ATP synthesis in skeletal muscle. In: Tipton CM, editor. ACSMs Advanced Exercise Physiology. Philadelphia (PA): Lippincott Williams and Wilkins;. p. 370-84.
47. Miller B. F. , Fattor J. A. , Jacobs K. A. , et al. (2002). Lactate and glucose interactions during rest and exercise in men: effect of exogenous lactate infusion. J. Physiol. 544:963-75.
48. Miller B. F. , Fattor J. A. , Jacobs K. A. et al. (2002). Metabolic and cardiores-piratory responses to "the lactate clamp". Am J Physiol Endocrinol Metab. 283:E889-98.
49. Nielsen H. B. , Clemmesen J. O. , Skak C. , Ott P. , Secher N. H. (2002). Attenuated hepatosplanchnic uptake of lactate during intense exercise in humans. J. Appl. Physiol. 92:1677-83.
50. Pagliassotti M. J. , Donovan C. M. (1990). Role of cell type in net lactate removal by skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 258:E635-642.
51. Pellerin L. (2005). How astrocytes feed hungry neurons. Mol. Neurobiol. 32:59-72.
52. Poole R. C. , Halestrap A. P. (1993). Transport of lactate and other mono-carboxylates across mammalian plasma membranes. Am. J. Physiol. Cell. Physiol. 264:C761-82.
53. Popinigis J. , Antosiewicz J. , Crimi M. , Lenaz G. , Wakabayashi T. (1991). Human skeletal muscle: participation of different metabolic activities in oxidation of L-lactate. Acta. Biochim. Pol. 38: 169-75.
54. Rasmussen H. N. , van Hall G. , Rasmussen U. F. (2002). Lactate dehydrogenase is not a mitochondrial enzyme in human and mouse vastus lateralis muscle. J. Physiol. 541:575-80.
55. Richter E. A. , Kiens B. , Saltin B. , Christensen N. J. , Savard G. (1988). Skeletal muscle glucose uptake during dynamic exercise in humans: role of muscle mass. Am. J. Physiol. Endocrinol. Metab. 254:E555-61.
56. Ring T. , Frische S. , Nielsen S. (2005). Clinical review: renal tubular acidosis-a physicochemical approach. Crit. Care. 9:573-80.
57. Robergs R. A. , Ghiasvand F. , Parker D. (2004). Biochemistry of exercise-induced metabolic acidosis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 287:R502-16.
58. Robergs R. A. , Ghiasvand F. , Parker D. (2005). Lingering construct of lactic acidosis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289: R904-10.
59. Robergs R. A. , Ghiasvand F. , Parker D. (2006). The wandering argument favoring a lactic acidosis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 291:R238-39.
60. Roef M. J. , de Meer K. , Kalhan S. C. , Straver H. , Berger R. , Reijngoud D. J. (2003). Gluconeogenesis in humans with induced hyperlactatemia during low-intensity exercise. Am. J. Physiol. Endocrinol. Metab. 284:E1162-71.
61. Roth D A , Brooks G A. (1990). Lactate transport is mediated by a membrane-bound carrier in rat skeletal muscle sarcolemmal vesicles. Arch. Biochem. Biophys. 279:377-85.
62. Sahlin K. , Fernstrom M. , Svensson M. , Tonkonogi M. (2002). No evidence of an intracellular lactate shuttle in rat skeletal muscle. J. Physiol. 541:569-74.
63. Sahlin K. , Harris R. C. , Nylind B. , Hultman E. (1976). Lactate content and pH in muscle obtained after dynamic exercise. Pflugers. Arch. 367:143-9.
64. Schurr A. (2006). Lactate: the ultimate cerebral oxidative energy substrate? J. Cereb. Blood Flow Metab. ;26:142-52.
65. Siggaard-Andersen O. (1963). The acid-base status of the blood. Scand. J. Clin. Lab. Invest. 15(Suppl. 70):1-134.
66. Siggaard-Andersen O, Fogh-Andersen N. (1995). Base excess or buffer base (strong ion difference) as measure of a non-respiratory acid-base disturbance. Acta Anaesthesiol. Scand. Suppl. 107:123-8.
67. Singer R. B. , Hastings A. B. (1948). An improved clinical method for the estimation of disturbances in the acid-base balance of human blood. Medicine (Baltimore). 27:223-42.
68. Stainsby W. N. , Brooks G. A. (1990). Control of lactic acid metabolism in contracting muscles and during exercise. Exerc. Sport. Sci. Rev. 18(l):29-63.
69. Stanley W. C. (1991). Myocardial lactate metabolism during exercise. Med. Sci. Sports. Exerc. 23(8):920-4.
70. Stanley W. C. , Gertz E. W. , Wisneski J. A. , Neese R. A. , Morris D. L. , Brooks G. A. (1986). Lactate extraction during net lactate release in legs of humans during exercise. J. Appl. Physiol. 60:1116-20.
71. Stanley W. C. , Wisneski J. A. , Gertz E. W. , Neese R. A. , Brooks G. A. (1988). Glucose and lactate interrelations during moderate-intensity exercise in humans. Metabolism. 37:850-8.
72. Stewart P. A. How to understand acid-base. (1981). In: A Quantitative Acid-Base Primer for Biology and Medicine. New York (NY): Elsevier. p. 1-186.
73. Szczesna-Kaczmarek A. (1990). L-lactate oxidation by skeletal muscle mitochondria. Int. J. Biochem. 22:617-20.
74. Van Hall G. , Jensen-Urstad M. , Rosdahl H. , Holmberg H. C. , Saltin B. , Calbet J. A. (2003). Leg and arm lactate and substrate kinetics during exercise. Am. J. Physiol. Endocrinol. Metab. 284:E193-205.
75. Vinnakota K. , Kemp M. L. , Kushmerick M. J. (2006). Dynamics of muscle glycogenolysis modeled with pH time course computation and pH-dependent reaction equilibria and enzyme kinetics. Biophys J. 91:1264-87.
76. Welch H. G. , Stainsby W. N. (1967). Oxygen debt in contracting dog skeletal muscle in situ. Respir Physiol. 3:229-42
77. Willis W. T. , Thompson A. , Messer J. I. , Thresher J. S. (2003). Vmax of mitochondrial electron shuttles in rat skeletal muscle and liver. Med Sci Sports Exerc. 35(5 suppl):S396.
78. Wooten E. W. (2003). Calculation of physiological acid-base parameters in multicompartment systems with application to human blood. J. Appl. Physiol. 95:2333-44.
79. Wooten E. W. (2004). Science review: quantitative acid-base physiology using the Stewart model. Crit. Care. 8:448-52.
80. Wooten E. W. (1998). Strong ion difference theory: more lessons from physical chemistry. Kidney Int. 54:1769.
81. Yoshida Y. , Holloway G. P. , Ljubicic V. , et al. (2007). Negligible direct lactate oxidation in subsarcolemmal and intermyofibrillar mitochondria obtained from red and white rat skeletal muscle. J. Physiol. 582:1317-35.